Identification and Characterization of Micrornas in Tree Peony During
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Eagle Rock Self-Guided Nature Trail
Eagle Rock Self-Guided sages. This evergreen plant has narrow dark green #6 - Dairy Creek, Camp San Luis and West Cuesta the spiny rolled edges. Like other oaks, the Coast leaves and orange-yellow flowers in spring and Ridge Vistas - Take a short break here and enjoy the Live Oak was a major food source for the Chumash Nature Trail summer. The young stems and leaves have been view to West Cuesta Ridge. Burned extensively in Indians. In early spring a golden glow may appear on used as salad greens by Indians. The crushed raw 1994 by the huge 41 these trees. If you take a branch covered with tiny Welcome to the Eagle Rock Self Guided Nature leaves and stems have been used to heal burns and Fire, the Los Padres catkins, you can see the yellow pollen fly on the Trail. This is a 1.8 mile trip to explore the flora, wounds. National Forest is breeze. This is the first step towards the tree making fauna and history of the Chorro Valley. This trail Coyote Bush (Baccharis making a strong acorns. offers great views of Chorro Valley and surrounding pilularis) is a common recovery. Many new hillsides. Take your time and enjoy your outdoor shrub of chaparral. It has seedlings have started to #9 - Dusky-footed Woodrat (Neotoma fuscipes) adventure. small leathery leaves 1/2 grow, including the Nests - As you look around you will see what seem If at the end of your adventure and you no longer to 1 inch long, with a Sergeant Cypress to be piles of debris up to 6 feet high. -

Plant Expedition to the Republic of Georgia
PLANT EXPEDITION TO THE REPUBLIC OF GEORGIA — CAUCASUS MOUNTAINS AUGUST 15 - SEPTEMBER 11, 2010 SPONSORED BY THE DANIEL F. AND ADA L. RICE FOUNDATION PLANT COLLECTING COLLABORATIVE (PCC) Chicago Botanic Garden Missouri Botanical Garden The Morton Arboretum New York Botanical Garden University of Minnesota Landscape Arboretum 1 Table of Contents Summary 3 Georgia’s Caucasus 4-6 Expedition, Expedition Route & Itinerary 7-10 Collaboration 11 Observations 12-13 Documentation 14 Institutional review 14-15 Acknowledgements 16 Maps of the Republic of Georgia and PCC member locations 17 Photo Gallery Collecting 18-19 Collections 20-24 Seed Processing 25 Landscapes 26-29 Transportation 30 Dining 31 People 32-33 Georgia Past and Present 34 Georgia News 35-36 Appendix I – Germplasm Collections Listed by Habit Appendix II – Germplasm Collections Listed Alphabetically Appendix III – Weed Risk Assessment Appendix IV – Field Notes 2 Summary With generous support from the Daniel F. and Ada L. Rice Foundation, Galen Gates and the Plant Collecting Collaborative (PCC) team made outstanding progress through an expedition in the Republic of Georgia. On this recent trip into the Caucasus Moun- tains, a record was set for the most collections made on any Chicago Botanic Garden and PCC expedition to date. The trip, door to door, was 26 days with field collecting most days; nearly every night‘s activity included seed cleaning. We made three hundred collections at 60 sites. Most were seeds from 246 types of trees, shrubs, and perennials, 14 were bulb taxa and four were in the form of perennial roots. Remarkably, 53 taxa are new to U.S. -

May 15, 2016 Passing Peony and Iris Plants on from Generation to Generation Annette Meyer Heisdorffer Daviess County Extension Agent for Horticulture
May 15, 2016 Passing Peony and Iris Plants on from Generation to Generation Annette Meyer Heisdorffer Daviess County Extension Agent for Horticulture After lunch on Mother’s Day, my mom and I surveyed her garden, especially the peonies. We both agreed that I needed to propagate her peonies and plant them in my garden. These are special, because I remember them growing in my grandmother’s garden. Peonies are commonly passed down from generation to generation. My goal is to someday share them with my twins. Our discussion included the irises, which are another heritage plant. Both plants are blooming beautifully in May and are spectacular in the garden. Information about these two plants will be provided in this article. Peony (Paeonia officinalis, Paeonia lactiflora, and hybrids) is a herbaceous perennial, which means at the end of the growing season it will die back to the ground. However, the plant returns year after year. Peonies grow best in full sun and well-drained soil. There are tree peonies (Paeonia suffruticosa) which have a woody stem, but those are not as common and require different growing conditions. The tree peony will not be discussed here. According to Dr. Rick Durham, Extension Specialist for Consumer Horticulture, peonies can be found in landscapes across Kentucky. Peonies have a long life span and are commonly grown in the garden. When planting the root, make sure it is not too deep. The eyes or bud should be just below the surface of the soil. If it is planted too deeply, the plants won't bloom. -

Peony Questions and Answers
Questions, Answers, and Comments Below are a list of questions and comments from the webinar chat feature. Black font corresponds to participant questions, comments, and answers. Some questions were answered during the webinar Colored font corresponds to comments and answers from the webinar speakers. Please visit https://cutflowers.ces.ncsu.edu/ to watch the webinar and hear questions answered during the presentations. Always do your own on-farm testing to validate a treatment or recommendation. Production Where are the gaps in production? Sept through late October and a small gap in January. Mention months again for harvest regions? Some higher elevation Alaska farms cut into early September In the central US we start in April in Arkansas and go until July in the UP When would we expect production for central/midwest? Late April in southern areas through early July in northern areas. About production in upstate NY and new England? New York and New England would see blooms in early June Is the peony bloom season for the southern hermisphere? Late October through January (often early January only). Number one top peony for cut flower production. (General ideas?) And why? in terms of bud production, which varieties tend to have the highest stem production? How many stems/plant should be expected? Possibly Sarah Bernhardt, due to having large fragrant flowers, high productivity, average but reliable vase life, reliable producer This really depends on the variety, the age of the plant, your management practices and environment. Jules Ellie has been our most prolific and highest gross earner Do you know how many chilling hours are demanded by variety? We don’t have chilling needs by variety. -

Fragrant Peonies
Self -guided tour for the University of Michigan, Nichols Arboretum Peony Garden Fragrant Peonies Peonies are often esteemed for their intoxicating fragrance. These scents range from sweet and rosy to citrusy and spicy. Surprisingly, not all peonies are fragrant. The double form white and pink peonies tend to be the most aromatic. Some semi-double and anemone formed peonies boast an attractive perfume as well. However, most single and red peonies do not have a scent—though there are a few exceptions. Below are some of the top-rated fragrant peonies in the Peony Garden. Smell and compare, then choose a favorite! 20 Splendida 26 27 18 La France 19 Richardson’s Grandiflora 24 25 14 Albert Crousse 17 John Richardson 13 16 Chestine Gowdy Do Tell 21 22 23 12 Avalanche 18 19 20 15 Sarah K. Thurlow 11 Bayadere 15 16 17 10 Mary Brand 12 13 14 7 James Kelway 9 La Perle 9 10 11 5 Nympheae 8 Mignon 6 7 8 4 Madame Emile Lemoine 3 Octavie Demay 3 4 5 6 Primevere 2 Madame Calot 1 2 1 Duchesse de Nemours Fragrant Peonies 1 1856 8 1908 15 1921 Duchesse de Nemours Mignon Sarah K. Thurlow This beautiful white This elegant peony This rose-scented peony has an blooms pink and peony blooms a intoxicating aroma. turns to flesh white pale pink and fades when fully open. It has to white as it opens. It has a 2 1856 a rich rose fragrance. smooth, rose-like center. Madame Calot 1886 9 16 1946 This early pink double peony produces many La Perle Do Tell flowers on sturdy stems This old-rose This peony and has a strong fragrance. -
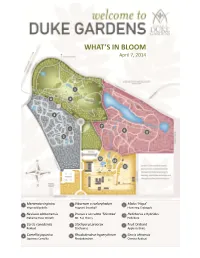
What's in Bloom
WHAT’S IN BLOOM April 7, 2014 5 4 6 2 7 1 9 8 3 12 10 11 1 Mertensia virginica 5 Viburnum x carlcephalum 9 Malus ‘Hopa’ Virginia Bluebells Fragrant Snowball Flowering Crabapple 2 Neviusia alabamensis 6 Prunus x serrulata ‘Shirotae’ 10 Helleborus x hybridus Alabama Snow Wreath Mt. Fuji Cherry Hellebore 3 Cercis canadensis 7 Stachyurus praecox 11 Fruit Orchard Redbud Stachyurus Apple cultivars 4 Camellia japonica 8 Rhododendron hyperythrum 12 Cercis chinensis Japanese Camellia Rhododendron Chinese Redbud WHAT’S IN BLOOM April 7, 2014 BLOMQUIST GARDEN OF NATIVE PLANTS Amelanchier arborea Common Serviceberry Sanguinaria canadensis Bloodroot Cornus florida Flowering Dogwood Stylophorum diphyllum Celandine Poppy Thalictrum thalictroides Rue Anemone Fothergilla major Fothergilla Trillium decipiens Chattahoochee River Trillium Hepatica nobilis Hepatica Trillium grandiflorum White Trillium Hexastylis virginica Wild Ginger Hexastylis minor Wild Ginger Trillium pusillum Dwarf Wakerobin Illicium floridanum Florida Anise Tree Trillium stamineum Blue Ridge Wakerobin Malus coronaria Sweet Crabapple Uvularia sessilifolia Sessileleaf Bellwort Mertensia virginica Virginia Bluebells Pachysandra procumbens Allegheny spurge Prunus americana American Plum DORIS DUKE CENTER GARDENS Camellia japonica Japanese Camellia Pulmonaria ‘Diana Clare’ Lungwort Cercis canadensis Redbud Prunus persica Flowering Peach Puschkinia scilloides Striped Squill Cercis chinensis Redbud Sanguinaria canadensis Bloodroot Clematis armandii Evergreen Clematis Spiraea prunifolia Bridalwreath -

Peony, Powdery Mildew (Erysiphe Polygoni)
Problem: Peony, Powdery Mildew (Erysiphe polygoni) Host Plants: Peony Description: Powdery mildew starts as individual spots that resemble snowflakes but rapidly coalesce to cover the entire leaf so that a plant look like it was dusted with flour. Though common on other plants such as lilac and bee balm, it has been relatively rare on peony until the last few years. Recommendations: Poor air movement and shade make the disease more likely. Growing peonies in full sun with good air movement will help minimize the disease. Fungicides can be effective if applied before infection has occurred. Therefore, heavily infected plants should not be treated as the treatment will be ineffective. Fortunately, the disease should cause no lasting damage to the plant. Remove and discard (or compost) infected plant material at the end of the season. Look for individual spots to appear the following spring and then apply a recommended fungicide before the disease has spread. Suggested fungicides include myclobutanil (Eagle, Spectracide Immunox, Monterey Fungi-Maxx, Fertilome F- Stop Lawn & Garden Fungicide), propiconazole (Banner MAXX, Fertilome Liquid Systemic Fungicide, Bonide Infuse Systemic Disease Control) or tebuconazole (BioAdvanced Disease Control for Roses, Flowers & Shrubs). References: 1. Powdery Mildew in the Flower Garden, University of Minnesota Extension 2. Peony Powdery Mildew, University of Illinois Extension, Home, Yard & Garden Pest Newsletter, July 16, 2010 Last Update: 1/16/2020 Brand names appearing in this publication are for product identification purposes only. No endorsement is intended, nor is criticism implied of similar products not mentioned. Kansas State University Agricultural Experiment Station and Cooperative Extension Service . -

The Tree Peonies
TI-IE NA.TIONA.L ~GA.rz J INE THE AMERICAN HORTICULTURAL SOCIETY, INC. 1600 Bladensburg Road, Northeast Washington 2, D. C. OFFICERS Presidellt: Dr. John L. Creech, Glenn Dale, :Ma ryland First Vice-Prcsidellt: Dr. Ezra ]. K raus, Corvalli s, Oregon Secolld Vice-Presiden t: I1{rs. Robert \"Toods Bli ss, vVashington, D. C. Secretary: Dr. Francis de Vos, Washington, D. C. Treasllrer: Miss Olive E. Vveatherell, Olean, New York Editor: Mr. B. Y. Morrison, Pass Christian, Mississipp i J1[ allagillg Editor: M r. James R. Harlow, Takoma Park, Maryland Editorial S tall : Miss May M. Blaine, Washington, D. C. Mr. Bernard T. Bridgers, Washington, D. C. Art Editor: Mr. Charl es C. Dickson, Kensington, Maryland DIRECTORS TerlJl s E xpirillg 1955 TerlJls E.,pir'ing 1956 Mrs. 'Mortim er J. Fox. Mount K isco, New Mr. Stuart Armstrong, Silver Spring, IVIa ry- Yo rk land lv[r. Frederic P. Lee, Bethesda, Maryland Dr. Fred O. Coe, Bethesda, Maryland Dr. Brian O. Mulligan, Seattl e, vVashington Mrs. Walter Douglas, Chauncey, New York Dr. F reeman A. vVeiss, Washington, D. C. Mrs. ]. Norman Henry, Gladwy ne, Penn- Dr. Donald vVyman, Jamaica P lain , Massa- sy lvania chusetts M rs. Arthur Hoyt Scott, Media, Pennsy l vallla HONORARY VICE-PRESIDENTS M r. James B. Craig Mr. George W. Peyton American Forestry Association American Peony Society 919 Seventee nth Street, Northwest Box No.1 \>\Tash in gton 6, D. C. Rapid an, V irgi ni a 'M r. Harry \ >\T . Dengler Mrs. Hermann G. P lace Holl y Society of America The Garden Club of America Maryland Extension Service 45 East 62nd Street Co ll ege Park, Maryland New York 21, New York Mr. -

Peony FREQUENTLY ASKED QUESTIONS FIRST THINGS FIRST
FREQUENTLY ASKED QUESTIONS WHEN CAN BLOOMS BE EXPECTED? In some cases you may get some flowers the first season in mid summer provided the temperatures do not turn into extreme heat too fast. If you do it will be nothing compared to the second season. Beginning the second spring season they produce impressive flowers. DO THE FLOWERS REALLY NEED STAKING? The flowers most often require some staking unless they are up against a wall and out of the wind. Often they grow to 4 feet tall and are too pretty to let be ruined in a storm. ARE ANTS ON THE BLOSSOMS A SIGN OF PESTS? No. Ants feed on the sweet sugary nectar of the flowers. Peonies are darn near pest free. DO PEONIES MAKE GOOD CUT FLOWERS? These make excellent cut flowers. Cut as much of the as you want! They drink a lot of water inside vase. Best time to cut is in the morning while they have buds that are cracking. CAN PEONIES BE DIVIDED? These roots will re-bloom every year. They can be subdivided in early spring after 5 years. Separate them making sure each section have at least 3 eyes or buds. Propagating can however make them skip blooming in the season they are divided. MY ROOTS ARE SLOW TO COME UP? Make sure that you did not plant them too deep. Pull out one and compare to the root picture inside this guide to make sure that they were planted with the top side up. They also need to be covered with only two to three inches of soil. -

Cool Season Annuals
Cool Season Annuals HORT 308/609 Assigned Readings for Plant List 6 Plant List 6 Spring 2020 Read the pages in your textbook associated with the family descriptions and individual taxa covered on Plant List 6 that was distributed in lab. These plant lists are also available on the course website All Text And Images Are Copyrighted By: Dr. Michael A. Arnold, Texas A&M University, Dept. Horticultural Sciences, College Station, TX 77843-2133 Cool season flowers Cool Season A bit of landscaping helps Annuals most any structure! • Tolerant of freezing to subfreezing temperatures – Suitable for use throughout winter in southern half of our region – Suitable for late fall and very early spring use in northern portions of the region • Provides off-season color in winter Cool season foliage Cool (Season) Thoughts Alcea rosea • Many species are derived from edible or Hollyhocks medicinal European species • Classic old-fashioned reseeding annual, • Plants utilized solely for foliage are biennial, or weak perennial more common than with other • Tolerates cold to USDA z. 5, but heat of z. 8 is tough seasonal annuals • Tall cool season annuals are infrequent, • Bold coarse textured foliage; rounded mound the or become tall only late in the season first year or winter and then stiffly upright in spring • Limited range of soil moisture is common • Most decline when day temperatures consistently exceed 80°F or night temperatures exceed 70°F • Mostly for detail designs, bedding, or seasonal containers • Miniature hibiscus-like flowers – Singles quaint, -

Flowering Perennials for Georgia Gardens
Flowering Perennials for Georgia Gardens Prepared by Paul A. Thomas, Extension Horticulturist Plants are classed according to their growth cycle as Some sources have suggested that perennials are not annuals, biennials or perennials. Annuals are short-lived well suited to the Southeast. This simply is not true. Many plants that complete their entire life cycle within one perennials perform exceedingly well in Georgia and the growing season. Biennials normally do not bloom until the Southeast in general. Not all perennials perform well here, second season, form seeds and then die. Perennials live but then not all perennials perform well in the Northeast from year to year, with varying bloom times. or even in England. Remember, much of the existing Perennials are also classed as woody (trees and shrubs literature regarding perennials is based on conditions that produce woody above-ground stems and branches that where cooler summer climates prevail. So exercise care in live from year to year) or herbaceous (plants that produce choosing plants well adapted for your particular area. comparatively soft tissues which often die back to ground Most perennials are completely winter hardy, although level at the end of the growing season). Herbaceous there are a few so-called tender perennials that are some- perennials persist by means of various underground times injured by low temperatures. The limiting factors in storage structures—bulbs, corms, tubers, tuberous stems, perennial adaptability in the Southeast are often heat toler- tuberous roots and crowns. ance and diseases that prevail in hot, humid climates. The distinction between annuals and perennials, woody and herbaceous, is not always sharply defined because climate influences growth potential. -

Section IX the STATE PAGES
Section IX THE STATE PAGES THE FOLLOWING section presents information on all the states of the United States and the District of Columbia; the commonwealths of Puerto Rico and the Northern Mariana Islands; the territories of American Samoa, Guam and the Virgin Islands; and the United Na tions trusteeships of the Federated States of Micronesia, the Marshall Islands and the Republic of Belau.* Included are listings of various executive officials, the justices of the supreme courts and officers of the legislatures. Lists of all officials are as of late 1981 or early 1982. Comprehensive listings of state legislators and other state officials appear in other publications of The Council of State Governments. Concluding each state listing are population figures and other statistics provided by the U.S. Bureau of the Census, based on the 1980 enumerafion. Preceding the state pages are three tables. The first lists the official names of states, the state capitols with zip codes and the telephone numbers of state central switchboards. The second table presents historical data on all the states, commonwealths and territories. The third presents a compilation of selected state statistics from the state pages. *The Northern Mariana Islands, the Federated States of Micronesia, the Marshall Islands and the Republic of Belau (formerly Palau) have been administered by the United Slates since July 18, 1947, as part of the Trust Territory of the Pacific Islands (TTPl), a trusteeship of the United Nations. The Northern Mariana Islands separated themselves from TTPI in March 1976 and now operate under a constitutional govern ment instituted January 9, 1978.