Museum Occurrence Data Predict Genetic Diversity in a Species-Rich Clade of Australian Lizards Supplementary Online Material
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reptile-Like Physiology in Early Jurassic Stem-Mammals
bioRxiv preprint doi: https://doi.org/10.1101/785360; this version posted October 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Reptile-like physiology in Early Jurassic stem-mammals Authors: Elis Newham1*, Pamela G. Gill2,3*, Philippa Brewer3, Michael J. Benton2, Vincent Fernandez4,5, Neil J. Gostling6, David Haberthür7, Jukka Jernvall8, Tuomas Kankanpää9, Aki 5 Kallonen10, Charles Navarro2, Alexandra Pacureanu5, Berit Zeller-Plumhoff11, Kelly Richards12, Kate Robson-Brown13, Philipp Schneider14, Heikki Suhonen10, Paul Tafforeau5, Katherine Williams14, & Ian J. Corfe8*. Affiliations: 10 1School of Physiology, Pharmacology & Neuroscience, University of Bristol, Bristol, UK. 2School of Earth Sciences, University of Bristol, Bristol, UK. 3Earth Science Department, The Natural History Museum, London, UK. 4Core Research Laboratories, The Natural History Museum, London, UK. 5European Synchrotron Radiation Facility, Grenoble, France. 15 6School of Biological Sciences, University of Southampton, Southampton, UK. 7Institute of Anatomy, University of Bern, Bern, Switzerland. 8Institute of Biotechnology, University of Helsinki, Helsinki, Finland. 9Department of Agricultural Sciences, University of Helsinki, Helsinki, Finland. 10Department of Physics, University of Helsinki, Helsinki, Finland. 20 11Helmholtz-Zentrum Geesthacht, Zentrum für Material-und Küstenforschung GmbH Germany. 12Oxford University Museum of Natural History, Oxford, OX1 3PW, UK. 1 bioRxiv preprint doi: https://doi.org/10.1101/785360; this version posted October 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 13Department of Anthropology and Archaeology, University of Bristol, Bristol, UK. 14Faculty of Engineering and Physical Sciences, University of Southampton, Southampton, UK. -

Lake Pinaroo Ramsar Site
Ecological character description: Lake Pinaroo Ramsar site Ecological character description: Lake Pinaroo Ramsar site Disclaimer The Department of Environment and Climate Change NSW (DECC) has compiled the Ecological character description: Lake Pinaroo Ramsar site in good faith, exercising all due care and attention. DECC does not accept responsibility for any inaccurate or incomplete information supplied by third parties. No representation is made about the accuracy, completeness or suitability of the information in this publication for any particular purpose. Readers should seek appropriate advice about the suitability of the information to their needs. © State of New South Wales and Department of Environment and Climate Change DECC is pleased to allow the reproduction of material from this publication on the condition that the source, publisher and authorship are appropriately acknowledged. Published by: Department of Environment and Climate Change NSW 59–61 Goulburn Street, Sydney PO Box A290, Sydney South 1232 Phone: 131555 (NSW only – publications and information requests) (02) 9995 5000 (switchboard) Fax: (02) 9995 5999 TTY: (02) 9211 4723 Email: [email protected] Website: www.environment.nsw.gov.au DECC 2008/275 ISBN 978 1 74122 839 7 June 2008 Printed on environmentally sustainable paper Cover photos Inset upper: Lake Pinaroo in flood, 1976 (DECC) Aerial: Lake Pinaroo in flood, March 1976 (DECC) Inset lower left: Blue-billed duck (R. Kingsford) Inset lower middle: Red-necked avocet (C. Herbert) Inset lower right: Red-capped plover (C. Herbert) Summary An ecological character description has been defined as ‘the combination of the ecosystem components, processes, benefits and services that characterise a wetland at a given point in time’. -

Cravens Peak Scientific Study Report
Geography Monograph Series No. 13 Cravens Peak Scientific Study Report The Royal Geographical Society of Queensland Inc. Brisbane, 2009 The Royal Geographical Society of Queensland Inc. is a non-profit organization that promotes the study of Geography within educational, scientific, professional, commercial and broader general communities. Since its establishment in 1885, the Society has taken the lead in geo- graphical education, exploration and research in Queensland. Published by: The Royal Geographical Society of Queensland Inc. 237 Milton Road, Milton QLD 4064, Australia Phone: (07) 3368 2066; Fax: (07) 33671011 Email: [email protected] Website: www.rgsq.org.au ISBN 978 0 949286 16 8 ISSN 1037 7158 © 2009 Desktop Publishing: Kevin Long, Page People Pty Ltd (www.pagepeople.com.au) Printing: Snap Printing Milton (www.milton.snapprinting.com.au) Cover: Pemberton Design (www.pembertondesign.com.au) Cover photo: Cravens Peak. Photographer: Nick Rains 2007 State map and Topographic Map provided by: Richard MacNeill, Spatial Information Coordinator, Bush Heritage Australia (www.bushheritage.org.au) Other Titles in the Geography Monograph Series: No 1. Technology Education and Geography in Australia Higher Education No 2. Geography in Society: a Case for Geography in Australian Society No 3. Cape York Peninsula Scientific Study Report No 4. Musselbrook Reserve Scientific Study Report No 5. A Continent for a Nation; and, Dividing Societies No 6. Herald Cays Scientific Study Report No 7. Braving the Bull of Heaven; and, Societal Benefits from Seasonal Climate Forecasting No 8. Antarctica: a Conducted Tour from Ancient to Modern; and, Undara: the Longest Known Young Lava Flow No 9. White Mountains Scientific Study Report No 10. -

Fowlers Gap Biodiversity Checklist Reptiles
Fowlers Gap Biodiversity Checklist ow if there are so many lizards then they should make tasty N meals for someone. Many of the lizard-eaters come from their Reptiles own kind, especially the snake-like legless lizards and the snakes themselves. The former are completely harmless to people but the latter should be left alone and assumed to be venomous. Even so it odern reptiles are at the most diverse in the tropics and the is quite safe to watch a snake from a distance but some like the Md rylands of the world. The Australian arid zone has some of the Mulga Snake can be curious and this could get a little most diverse reptile communities found anywhere. In and around a disconcerting! single tussock of spinifex in the western deserts you could find 18 species of lizards. Fowlers Gap does not have any spinifex but even he most common lizards that you will encounter are the large so you do not have to go far to see reptiles in the warmer weather. Tand ubiquitous Shingleback and Central Bearded Dragon. The diversity here is as astonishing as anywhere. Imagine finding six They both have a tendency to use roads for passage, warming up or species of geckos ranging from 50-85 mm long, all within the same for display. So please slow your vehicle down and then take evasive genus. Or think about a similar diversity of striped skinks from 45-75 action to spare them from becoming a road casualty. The mm long! How do all these lizards make a living in such a dry and Shingleback is often seen alone but actually is monogamous and seemingly unproductive landscape? pairs for life. -

Expert Report of Professor Woinarski
NOTICE OF FILING This document was lodged electronically in the FEDERAL COURT OF AUSTRALIA (FCA) on 18/01/2019 3:23:32 PM AEDT and has been accepted for filing under the Court’s Rules. Details of filing follow and important additional information about these are set out below. Details of Filing Document Lodged: Expert Report File Number: VID1228/2017 File Title: FRIENDS OF LEADBEATER'S POSSUM INC v VICFORESTS Registry: VICTORIA REGISTRY - FEDERAL COURT OF AUSTRALIA Dated: 18/01/2019 3:23:39 PM AEDT Registrar Important Information As required by the Court’s Rules, this Notice has been inserted as the first page of the document which has been accepted for electronic filing. It is now taken to be part of that document for the purposes of the proceeding in the Court and contains important information for all parties to that proceeding. It must be included in the document served on each of those parties. The date and time of lodgment also shown above are the date and time that the document was received by the Court. Under the Court’s Rules the date of filing of the document is the day it was lodged (if that is a business day for the Registry which accepts it and the document was received by 4.30 pm local time at that Registry) or otherwise the next working day for that Registry. No. VID 1228 of 2017 Federal Court of Australia District Registry: Victoria Division: ACLHR FRIENDS OF LEADBEATER’S POSSUM INC Applicant VICFORESTS Respondent EXPERT REPORT OF PROFESSOR JOHN CASIMIR ZICHY WOINARSKI Contents: 1. -

APPENDICES Appendix A. Fauna Species Recorded on Brigalow Research Station. Introduced Species Denoted by *, Vulnerable
APPENDICES Appendix A. Fauna species recorded on Brigalow Research Station. Introduced species denoted by *, vulnerable 'V', rare 'IR'. Scientific Name Common Name 1. Tachyglossus aculeatus Short-beaked echidna 2. Planigale maculata Common planigale 3. Sminthopsis macroura Stripe-faced dunnart 4. Isoodon macrourus Northern brown (Giant brindled) bandicoot 5. Phascolarctos cinereus Koala 6. Petaurus breviceps Sugar glider 7. Petaurus norfolcensis Squirrel glider 8. Petauroides volans Greater glider 9. Trichosurus vulpecula Common brushtail possum 10. Aepyprymnus rufescens Rufous bettong 11. Macropus dorsalis Black-striped wallaby 12. Macropus giganteus Eastern grey kangaroo 13. Macropus parryi Whiptail wallaby 14. Wallabia bicolor Swamp wallaby 15. Pteropus scapulatus Little red flying fox 16. Hydromys chrysogaster Water rat 17. Mus musculus* House mouse 18. Pseudomys delicatulus Delicate mouse 19. Canis lupus dingo Dingo 20. Canis sp.* Domestic dog 21. Vulpes vulpes* Fox 22. Felis catus* Cat 23. Oryctolagus cuniculus* Rabbit 24. Lepus capensis* Hare 25. Equus sp. * Domestic horse 26. Sus scrofa* Feral pig 27. Bus sp. * Domestic cow 28. Capra hircus* Domestic goat 29. Limnodynastes ornatus Ornate burrowing frog 30. Limnodynastes salmini Salmon-striped frog 31. Limnodynastes tasmaniensis Spotted marsh frog 32. Limnodynastes terraereginae Northern (Banjo) pobblebonk frog 33. Uperoleia rugosa Wrinkled (Eastern Burrowing) toadlet 34. Cyclorana brevipes Short-footed (Blotched) waterholding frog 35. Cyclorana novaehollandiae New Holland frog 36. Litoria alboguttata Green-striped frog 37. Litoria caerulea Green tree frog 38. Litoria fallax Eastern sedge frog 39. Litoria inermis Floodplain frog 40. Litoria latopalmata Broad-palmed frog 41. Litoria peronii Emerald spotted (Peron's tree) frog 42. Litoria rubella Desert tree frog 43. Bufo marinus* Cane toad 44. -

Literature Cited in Lizards Natural History Database
Literature Cited in Lizards Natural History database Abdala, C. S., A. S. Quinteros, and R. E. Espinoza. 2008. Two new species of Liolaemus (Iguania: Liolaemidae) from the puna of northwestern Argentina. Herpetologica 64:458-471. Abdala, C. S., D. Baldo, R. A. Juárez, and R. E. Espinoza. 2016. The first parthenogenetic pleurodont Iguanian: a new all-female Liolaemus (Squamata: Liolaemidae) from western Argentina. Copeia 104:487-497. Abdala, C. S., J. C. Acosta, M. R. Cabrera, H. J. Villaviciencio, and J. Marinero. 2009. A new Andean Liolaemus of the L. montanus series (Squamata: Iguania: Liolaemidae) from western Argentina. South American Journal of Herpetology 4:91-102. Abdala, C. S., J. L. Acosta, J. C. Acosta, B. B. Alvarez, F. Arias, L. J. Avila, . S. M. Zalba. 2012. Categorización del estado de conservación de las lagartijas y anfisbenas de la República Argentina. Cuadernos de Herpetologia 26 (Suppl. 1):215-248. Abell, A. J. 1999. Male-female spacing patterns in the lizard, Sceloporus virgatus. Amphibia-Reptilia 20:185-194. Abts, M. L. 1987. Environment and variation in life history traits of the Chuckwalla, Sauromalus obesus. Ecological Monographs 57:215-232. Achaval, F., and A. Olmos. 2003. Anfibios y reptiles del Uruguay. Montevideo, Uruguay: Facultad de Ciencias. Achaval, F., and A. Olmos. 2007. Anfibio y reptiles del Uruguay, 3rd edn. Montevideo, Uruguay: Serie Fauna 1. Ackermann, T. 2006. Schreibers Glatkopfleguan Leiocephalus schreibersii. Munich, Germany: Natur und Tier. Ackley, J. W., P. J. Muelleman, R. E. Carter, R. W. Henderson, and R. Powell. 2009. A rapid assessment of herpetofaunal diversity in variously altered habitats on Dominica. -
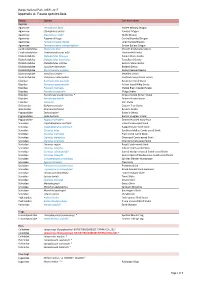
Report-Mungo National Park-Appendix A
Mungo National Park, NSW, 2017 Appendix A: Fauna species lists Family Species Common name Reptiles Agamidae Ctenophorus fordi Mallee Military Dragon Agamidae Ctenophorus pictus Painted Dragon Agamidae Diporiphora nobbi Nobbi Dragon Agamidae Pogona vitticeps Central Bearded Dragon Agamidae Tympanocryptis lineata Lined Earless Dragon Agamidae Tympanocryptis tetraporophora Eyrean Earless Dragon Carphodactylidae Nephrurus levis Smooth Knob-tailed Gecko Carphodactylidae Underwoodisaurus milii Thick-tailed Gecko Diplodactylidae Diplodactylus furcosus Ranges Stone Gecko Diplodactylidae Diplodactylus tessellatus Tessellated Gecko Diplodactylidae Diplodactylus vittatus Eastern Stone Gecko Diplodactylidae Lucasium damaeum Beaded Gecko Diplodactylidae Rhynchoedura ormsbyi Eastern Beaked Gecko Diplodactylidae Strophurus elderi ~ Jewelled Gecko Diplodactylidae Strophurus intermedius Southern Spiny-tailed Gecko Elapidae Brachyurophis australis Australian Coral Snake Elapidae Demansia psammophis Yellow-faced Whip Snake Elapidae Parasuta nigriceps Mallee Black-headed Snake Elapidae Pseudechis australis Mulga Snake Elapidae Pseudonaja aspidorhyncha * Strap-snouted Brown Snake Elapidae Pseudonaja textilis Eastern Brown Snake Elapidae Suta suta Curl Snake Gekkonidae Gehyra versicolor Eastern Tree Gecko Gekkonidae Heteronotia binoei Bynoe's Gecko Pygopodidae Delma butleri Butler's Delma Pygopodidae Lialis burtonis Burton's Legless Lizard Pygopodidae Pygopus schraderi Eastern Hooded Scaly-foot Scincidae Cryptoblepharus australis Inland Snake-eyed Skink Scincidae -

(Reptilia: Squamata: Scincidae) Species Group and a New Species of Immediate Conservation Concern in the Southwestern Australian Biodiversity Hotspot
Zootaxa 3390: 1–18 (2012) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2012 · Magnolia Press ISSN 1175-5334 (online edition) Molecular phylogeny and morphological revision of the Ctenotus labillardieri (Reptilia: Squamata: Scincidae) species group and a new species of immediate conservation concern in the southwestern Australian biodiversity hotspot GEOFFREY M. KAY & J. SCOTT KEOGH 1 Division of Evolution, Ecology and Genetics, Research School of Biology, The Australian National University, Canberra, ACT 0200, Australia 1 Corresponding author. E-mail: [email protected] Abstract Ctenotus is the largest and most diverse genus of skinks in Australia with at least 97 described species. We generated large mitochondrial and nuclear DNA data sets for 70 individuals representing all available species in the C. labillardieri species- group to produce the first comprehensive phylogeny for this clade. The widespread C. labillardieri was sampled extensively to provide the first detailed phylogeographic data set for a reptile in the southwestern Australian biodiversity hotspot. We supplemented our molecular data with a comprehensive morphological dataset for the entire group, and together these data are used to revise the group and describe a new species. The morphologically highly variable species C. labillardieri comprises seven well-supported genetic clades that each occupy distinct geographic regions. The phylogeographic patterns observed in this taxon are consistent with studies of frogs, plants and invertebrates, adding strength to emerging biogeographic hypotheses in this iconic region. The species C. catenifer, C. youngsoni, and C. gemmula are well supported, and despite limited sampling both C. catenifer and C. gemmula show substantial genetic structure. -

No Evidence for the 'Rate-Of-Living' Theory Across the Tetrapod Tree of Life
Received: 23 June 2019 | Revised: 30 December 2019 | Accepted: 7 January 2020 DOI: 10.1111/geb.13069 RESEARCH PAPER No evidence for the ‘rate-of-living’ theory across the tetrapod tree of life Gavin Stark1 | Daniel Pincheira-Donoso2 | Shai Meiri1,3 1School of Zoology, Faculty of Life Sciences, Tel Aviv University, Tel Aviv, Israel Abstract 2School of Biological Sciences, Queen’s Aim: The ‘rate-of-living’ theory predicts that life expectancy is a negative function of University Belfast, Belfast, United Kingdom the rates at which organisms metabolize. According to this theory, factors that accel- 3The Steinhardt Museum of Natural History, Tel Aviv University, Tel Aviv, Israel erate metabolic rates, such as high body temperature and active foraging, lead to organismic ‘wear-out’. This process reduces life span through an accumulation of bio- Correspondence Gavin Stark, School of Zoology, Faculty of chemical errors and the build-up of toxic metabolic by-products. Although the rate- Life Sciences, Tel Aviv University, Tel Aviv, of-living theory is a keystone underlying our understanding of life-history trade-offs, 6997801, Israel. Email: [email protected] its validity has been recently questioned. The rate-of-living theory has never been tested on a global scale in a phylogenetic framework, or across both endotherms and Editor: Richard Field ectotherms. Here, we test several of its fundamental predictions across the tetrapod tree of life. Location: Global. Time period: Present. Major taxa studied: Land vertebrates. Methods: Using a dataset spanning the life span data of 4,100 land vertebrate spe- cies (2,214 endotherms, 1,886 ectotherms), we performed the most comprehensive test to date of the fundamental predictions underlying the rate-of-living theory. -

Fitzroy, Queensland
Biodiversity Summary for NRM Regions Species List What is the summary for and where does it come from? This list has been produced by the Department of Sustainability, Environment, Water, Population and Communities (SEWPC) for the Natural Resource Management Spatial Information System. The list was produced using the AustralianAustralian Natural Natural Heritage Heritage Assessment Assessment Tool Tool (ANHAT), which analyses data from a range of plant and animal surveys and collections from across Australia to automatically generate a report for each NRM region. Data sources (Appendix 2) include national and state herbaria, museums, state governments, CSIRO, Birds Australia and a range of surveys conducted by or for DEWHA. For each family of plant and animal covered by ANHAT (Appendix 1), this document gives the number of species in the country and how many of them are found in the region. It also identifies species listed as Vulnerable, Critically Endangered, Endangered or Conservation Dependent under the EPBC Act. A biodiversity summary for this region is also available. For more information please see: www.environment.gov.au/heritage/anhat/index.html Limitations • ANHAT currently contains information on the distribution of over 30,000 Australian taxa. This includes all mammals, birds, reptiles, frogs and fish, 137 families of vascular plants (over 15,000 species) and a range of invertebrate groups. Groups notnot yet yet covered covered in inANHAT ANHAT are notnot included included in in the the list. list. • The data used come from authoritative sources, but they are not perfect. All species names have been confirmed as valid species names, but it is not possible to confirm all species locations. -

Threatened Species Scientific Committee's Advice for the Wetlands and Inner Floodplains of the Macquarie Marshes
Threatened Species Scientific Committee’s Advice for the Wetlands and inner floodplains of the Macquarie Marshes 1. The Threatened Species Scientific Committee (the Committee) was established under the Environment Protection and Biodiversity Conservation Act 1999 (EPBC Act) and has obligations to present advice to the Minister for the Environment, Heritage and Water (the Minister) in relation to the listing and conservation of threatened ecological communities, including under sections 189, 194N and 266B of the EPBC Act. 2. The Committee provided this advice on the Wetlands and inner floodplains of the Macquarie Marshes ecological community to the Minister in June 2013. 3. A copy of the draft advice for this ecological community was made available for expert and public comment for a minimum of 30 business days. The Committee and Minister had regard to all public and expert comment that was relevant to the consideration of the ecological community. 4. This advice has been developed based on the best available information at the time it was assessed: this includes scientific literature, government reports, extensive advice from consultations with experts, and existing plans, records or management prescriptions for this ecological community. 5. This ecological community was listed as critically enadangered from 13 August 2013 to 11 December 2013. The listing was disallowed on 11 December 2013. It is no longer a matter of National Environmental Significance under the EPBC Act. Page 1 of 99 Threatened Species Scientific Committee’s Advice: