210598Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

COPD: Treatment Updates and Transitions of Care
COPD: Treatment Updates and Transitions of Care PRESENTED AS A LIVE WEBINAR ON-DEMAND ACTIVITY Thursday, May 21, 2020 Release date: 5/31/2020 1:00 p.m. - 2:00 p.m. Expiration date: 5/31/2023 FACULTY Dennis M. Williams, Pharm.D., BCPS, AE-C, FASHP, FCCP, FAPhA Associate Professor Division of Pharmacotherapy and Experimental Therapeutics UNC Eshelman School of Pharmacy University of North Carolina Chapel Hill, North Carolina Bradley Drummond, M.D., MHS Associate Professor of Medicine University of North Carolina at Chapel Hill Chapel Hill, North Carolina View faculty bios at https://www.copdcare.org/faculty-bios.php ASHP FINANCIAL RELATIONSHIP DISCLOSURE STATEMENT Planners, presenters, reviewers, ASHP staff, and others with an opportunity to control CE content are required to disclose relevant financial relationships with ACCME-defined commercial interests. All actual conflicts of interest have been resolved prior to the continuing education activity taking place. ASHP will disclose financial relationship information prior to the beginning of the activity. A relevant financial relationship is a defined as a financial relationship between an individual (or spouse/partner) in control of content and a commercial interest, in any amount, in the past 12 months, and products and/or services of the commercial interest (with which they have the financial relationship) are related to the continuing education activity. An ACCME-defined commercial interest is any entity producing, marketing re-selling, or distributing healthcare goods or services consumed by, or used on, patients. The ACCME does not consider providers of clinical serve directly to patients to be commercial interests—unless the provider of clinical service is owned, or controlled by, an ACCME-defined commercial interest. -

COPD Agents Review – October 2020 Page 2 | Proprietary Information
COPD Agents Therapeutic Class Review (TCR) October 1, 2020 No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, digital scanning, or via any information storage or retrieval system without the express written consent of Magellan Rx Management. All requests for permission should be mailed to: Magellan Rx Management Attention: Legal Department 6950 Columbia Gateway Drive Columbia, Maryland 21046 The materials contained herein represent the opinions of the collective authors and editors and should not be construed to be the official representation of any professional organization or group, any state Pharmacy and Therapeutics committee, any state Medicaid Agency, or any other clinical committee. This material is not intended to be relied upon as medical advice for specific medical cases and nothing contained herein should be relied upon by any patient, medical professional or layperson seeking information about a specific course of treatment for a specific medical condition. All readers of this material are responsible for independently obtaining medical advice and guidance from their own physician and/or other medical professional in regard to the best course of treatment for their specific medical condition. This publication, inclusive of all forms contained herein, is intended to be educational in nature and is intended to be used for informational purposes only. Send comments and suggestions to [email protected]. October 2020 -

Efficacy of Revefenacin, a Long-Acting Muscarinic Antagonist for Nebulized Therapy, in Patients with Markers of More Severe COPD: a Post Hoc Subgroup Analysis James F
Donohue et al. BMC Pulmonary Medicine (2020) 20:134 https://doi.org/10.1186/s12890-020-1156-4 RESEARCH ARTICLE Open Access Efficacy of revefenacin, a long-acting muscarinic antagonist for nebulized therapy, in patients with markers of more severe COPD: a post hoc subgroup analysis James F. Donohue1, Edward Kerwin2, Chris N. Barnes3, Edmund J. Moran3, Brett Haumann3 and Glenn D. Crater3* Abstract Background: Revefenacin, a once-daily, long-acting muscarinic antagonist delivered via standard jet nebulizer, increased trough forced expiratory volume in 1 s (FEV1) in patients with moderate to very severe chronic obstructive pulmonary disease (COPD) in prior phase 3 trials. We evaluated the efficacy of revefenacin in patients with markers of more severe COPD. Methods: A post hoc subgroup analysis of two replicate, randomized, phase 3 trials was conducted over 12 weeks. Endpoints included least squares change from baseline in trough FEV1, St. George’s Respiratory Questionnaire (SGRQ) responders, and transition dyspnea index (TDI) responders at Day 85. This analysis included patient subgroups at high risk for COPD exacerbations and compared patients who received revefenacin 175 μg and placebo: severe and very severe airflow limitation (percent predicted FEV1 30%–< 50% and < 30%), 2011 Global Initiative for Chronic Obstructive Lung Disease (GOLD) D, reversibility (≥ 12% and ≥ 200 mL increase in FEV1)to short-acting bronchodilators, concurrent use of long-acting β agonists and/or inhaled corticosteroids, older age (> 65 and > 75 years), and comorbidity risk factors. Results: Revefenacin demonstrated significant improvements in FEV1 versus placebo at Day 85 among the intention-to-treat (ITT) population and all subgroups. -

FDA Briefing Document Pulmonary-Allergy Drugs Advisory Committee Meeting
FDA Briefing Document Pulmonary-Allergy Drugs Advisory Committee Meeting August 31, 2020 sNDA 209482: fluticasone furoate/umeclidinium/vilanterol fixed dose combination to reduce all-cause mortality in patients with chronic obstructive pulmonary disease NDA209482/S-0008 PADAC Clinical and Statistical Briefing Document Fluticasone furoate/umeclidinium/vilanterol fixed dose combination for all-cause mortality DISCLAIMER STATEMENT The attached package contains background information prepared by the Food and Drug Administration (FDA) for the panel members of the advisory committee. The FDA background package often contains assessments and/or conclusions and recommendations written by individual FDA reviewers. Such conclusions and recommendations do not necessarily represent the final position of the individual reviewers, nor do they necessarily represent the final position of the Review Division or Office. We have brought the supplemental New Drug Application (sNDA) 209482, for fluticasone furoate/umeclidinium/vilanterol, as an inhaled fixed dose combination, for the reduction in all-cause mortality in patients with COPD, to this Advisory Committee in order to gain the Committee’s insights and opinions, and the background package may not include all issues relevant to the final regulatory recommendation and instead is intended to focus on issues identified by the Agency for discussion by the advisory committee. The FDA will not issue a final determination on the issues at hand until input from the advisory committee process has been considered -

HIM.PA.153 Inhaled Agents for Asthma and COPD
Clinical Policy: Inhaled Agents for Asthma and COPD Reference Number: HIM.PA.153 Effective Date: 03.01.21 Last Review Date: 02.21 Line of Business: HIM Revision Log See Important Reminder at the end of this policy for important regulatory and legal information. Description The following are inhaled agents for asthma and/or chronic obstructive pulmonary disease (COPD) requiring prior authorization: • Short acting beta-2 agonist (SABA): albuterol (ProAir® Digihaler®), levalbuterol (Xopenex® HFA, Xopenex® inhalation solution) • Inhaled corticosteroid (ICS): budesonide (Pulmicort Respules®)*, ciclesonide (Alvesco®), fluticasone (Armonair® Digihaler™), mometasone (Asmanex® HFA, Asmanex® Twisthaler®) • Long acting beta-2 agonist (LABA): arformoterol (Brovana®), formoterol (Perforormist) • Long acting muscarinic antagonist (LAMA): aclidinium bromide (Tudorza® Pressair®), glycopyrrolate (Seebri™ Neohaler®, Lonhala® Magnair®), revefenacin (Yupelri®) • Combination ICS/LABA: budesonide/formoterol (Symbicort®)*, fluticasone/salmeterol (Advair Diskus®*, AirDuo® Digihaler™, AirDuo® RespiClick®), mometasone/formoterol (Dulera®) • Combination LABA/LAMA: aclidnium/formoterol (Duaklir® Pressair®), indacaterol/glycopyrrolate (Utibron™ Neohaler®), tiotropium/olodaterol (Stiolto® Respimat®) • Combination ICS/LAMA/LABA: budesonide/glycopyrrolate/formoterol (Breztri Aerosphere™) _______________ *Generic agents do not require prior authorization. FDA Approved Indication(s) ProAir Digihaler and Xopenex are indicated for the treatment or prevention of bronchospasm -

Clinical Reporting System (BGP) Version 21.1
RESOURCE AND PATIENT MANAGEMENT SYSTEM Clinical Reporting System (BGP) User Manual Version 21.1 July 2021 Office of Information Technology Division of Information Technology Clinical Reporting System (BGP) Version 21.1 Table of Contents 1.0 Introduction......................................................................................................... 1 1.1 Key Changes in v21.1 .............................................................................. 1 1.1.1 Logic Changes to National GPRA/GPRAMA Report Measures............. 1 1.1.2 Key Logic Changes to Non-GPRA Measures ........................................ 2 1.1.3 Additional Key Enhancements and Revisions ....................................... 3 2.0 Orientation .......................................................................................................... 4 3.0 Clinical Reporting System ................................................................................. 5 3.1 Clinical Performance Assessment and GPRA ......................................... 5 3.1.1 What Is GPRA? ..................................................................................... 5 3.1.2 Clinical Performance Measures ............................................................. 7 3.1.3 Comparing Ourselves to National Guidelines ........................................ 8 3.2 CRS Overview ......................................................................................... 8 3.2.1 How Does CRS Work? .......................................................................... 9 3.2.2 -

A 28-Day, Randomized, Double-Blind, Placebo-Controlled, Parallel Group Study of Nebulized Revefenacin in Patients with Chronic Obstructive Pulmonary Disease
A 28-day, Randomized, Double-Blind, Placebo-Controlled, Parallel Group Study of Nebulized Revefenacin in Patients With Chronic Obstructive Pulmonary Disease BACKGROUND: • Long-acting bronchodilators are an important part of therapy for patients with COPD. Monotherapy with a long- acting muscarinic antagonist (LAMA) is considered first-line in some patients. Revefenacin is a new once daily nebulizer LAMA therapy approved for maintenance treatment of COPD. • Sometimes, other dosage forms (metered-dose and dry powder inhalers) are not easy to use, especially in older patients or those with cognitive impairment. These factors may cause inaccurate dosing, and poor adherence leading to undesirable clinical outcomes. OBJECTIVE • To evaluate the safety, efficacy, and tolerability of revefenacin in patients with COPD. METHODS • Design: randomized, double-blind, placebo-controlled, multiple-dose, parallel-group study conducted at 41 US centers within a 4 month period in 2014. 70 participants were assigned to each of 5 treatment groups: o Group 1: placebo o Group 3: 88 mcg o Group 5: 350 mcg o Group 2: 44 mcg revefenacin revefenacin revefenacin o Group 4: 175 mcg revefenacin • Exclusion: patients who had a >12% change or >200 mL increase in FEV1 after placebo inhalation. o LABA or LAMA use was not allowed during the study o Patients using LABA/ICS therapy ICS monotherapy was allowed (LABA component discontinued) o SABA therapy was allowed except 6 hours before the initial spirometry assessment at each study visit • Inclusion: men and women ages 40 to 75 with moderate to severe COPD, current or former smoker (>10 pack- years), with a post-ipratropium bromide FEV1:FVC <0.7, FEV1 30-80% that of the predicted normal value after withholding short-acting bronchodilators for at least 6 hours and long-acting bronchodilators for at least 14 days. -

Inhaled Glycopyrrolate for the Treatment of Chronic Obstructive Pulmonary Disease
Journal name: International Journal of COPD Article Designation: Review Year: 2018 Volume: 13 International Journal of COPD Dovepress Running head verso: Tashkin and Gross Running head recto: Inhaled glycopyrrolate for COPD open access to scientific and medical research DOI: 162646 Open Access Full Text Article REVIEW Inhaled glycopyrrolate for the treatment of chronic obstructive pulmonary disease 1 Donald P Tashkin Abstract: Long-acting muscarinic antagonists (LAMAs), along with long-acting β2-agonists Nicholas J Gross2 (LABAs), are the mainstay for treatment of patients with COPD. Glycopyrrolate, or glycopyr- ronium bromide, like other LAMAs, inhibits parasympathetic nerve impulses by selectively 1Department of Medicine, David Geffen School of Medicine at UCLA, blocking the binding of acetylcholine to muscarinic receptors. Glycopyrrolate is unusual in that Los Angeles, CA, USA; 2Department it preferentially binds to M over M muscarinic receptors, thereby specifically targeting the of Medicine, University Medical 3 2 Research LLC, Saint Francis Hospital primary muscarinic receptor responsible for bronchoconstriction occurring in COPD. Inhaled and Medical Center, Hartford, glycopyrrolate is slowly absorbed from the lungs and rapidly eliminated from the bloodstream, CT, USA most likely by renal excretion in its unmetabolized form, limiting the potential for systemic adverse events. Inhaled glycopyrrolate is a fast-acting, efficacious treatment option for patients with moderate–severe COPD. It improves lung function, reduces the risk of exacerbations, and alleviates the symptoms of breathlessness, which in turn may explain the improvement seen in For personal use only. patients’ quality of life. Inhaled formulations containing glycopyrrolate are well tolerated, and despite being an anticholinergic, few cardiovascular-related events have been reported. -
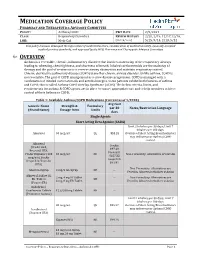
Asthma COPD and Asthma-COPD Overlap Syndrome (ACOS)
MEDICATION COVERAGE POLICY PHARMACY AND THERAPEUTICS ADVISORY COMMITTEE POLICY: Asthma/COPD P&T DATE 2/9/2021 CLASS: Respiratory Disorders REVIEW HISTORY 2/20, 2/19, 12/17,12/16, LOB: Medi-Cal (MONTH/YEAR) 5/15, 9/14, 2/13, 5/12 This policy has been developed through review of medical literature, consideration of medical necessity, generally accepted medical practice standards, and approved by the HPSJ Pharmacy and Therapeutic Advisory Committee OVERVIEW Asthma is a reversible, chronic, inflammatory disorder that involves narrowing of the respiratory airways leading to wheezing, chest tightness, and shortness of breath. Inhaled corticosteroids are the mainstay of therapy and the goal of treatment is to reverse airway obstruction and maintain respiratory control. Chronic obstructive pulmonary disease (COPD) is another chronic airway disorder. Unlike asthma, COPD is not reversible. The goal of COPD management is to slow disease progression. COPD is managed with a combination of inhaled corticosteroids and anticholinergics. Some patients exhibit both features of asthma and COPD; this is called Asthma-COPD Overlap Syndrome (ACOS). The below criteria, limits, and requirements for asthma & COPD agents are in place to ensure appropriate use and to help members achieve control of their Asthma or COPD. Table 1: Available Asthma/COPD Medications (Current as of 1/2020) Avg Cost Generic Name Strength & Formulary per 30 Notes/Restriction Language (Brand Name) Dosage form Limits days Single Agents Short Acting Beta Agonist (SABA) Limit 2 inhalers per 30 days; Limit 7 inhalers per 180 days. Albuterol 90 mcg/act QL $53.28 Overuse of Short Acting Bronchodilators may indicate poor Asthma/COPD control. -

BMJ Open Is Committed to Open Peer Review. As Part of This Commitment We Make the Peer Review History of Every Article We Publish Publicly Available
BMJ Open: first published as 10.1136/bmjopen-2018-027935 on 5 May 2019. Downloaded from BMJ Open is committed to open peer review. As part of this commitment we make the peer review history of every article we publish publicly available. When an article is published we post the peer reviewers’ comments and the authors’ responses online. We also post the versions of the paper that were used during peer review. These are the versions that the peer review comments apply to. The versions of the paper that follow are the versions that were submitted during the peer review process. They are not the versions of record or the final published versions. They should not be cited or distributed as the published version of this manuscript. BMJ Open is an open access journal and the full, final, typeset and author-corrected version of record of the manuscript is available on our site with no access controls, subscription charges or pay-per-view fees (http://bmjopen.bmj.com). If you have any questions on BMJ Open’s open peer review process please email [email protected] http://bmjopen.bmj.com/ on September 26, 2021 by guest. Protected copyright. BMJ Open BMJ Open: first published as 10.1136/bmjopen-2018-027935 on 5 May 2019. Downloaded from Treatment of stable chronic obstructive pulmonary disease: a protocol for a systematic review and evidence map Journal: BMJ Open ManuscriptFor ID peerbmjopen-2018-027935 review only Article Type: Protocol Date Submitted by the 15-Nov-2018 Author: Complete List of Authors: Dobler, Claudia; Mayo Clinic, Evidence-Based Practice Center, Robert D. -

YUPELRI (Revefenacin) Inhalation Solution
patients to contact a healthcare provider immediately if HIGHLIGHTS OF PRESCRIBING symptoms occur. (5.3) INFORMATION • Worsening of urinary retention may occur. Use with caution in patients with prostatic hyperplasia or bladder-neck These highlights do not include all the information needed to obstruction and instruct patients to contact a healthcare use YUPELRI™ (revefenacin) inhalation solution safely and provider immediately if symptoms occur. (5.4) effectively. See full prescribing information for YUPELRI (revefenacin) inhalation solution. • Immediate hypersensitivity reactions may occur. If such a reaction occurs, therapy with YUPELRI should be stopped at YUPELRI (revefenacin) inhalation solution, for oral inhalation once and alternative treatments should be considered. (5.5) Initial U.S. Approval: 2018 ------------------------ADVERSE REACTIONS----------------------- ----------------------INDICATIONS AND USAGE------------------- Most common adverse reactions (incidence greater than or equal to YUPELRI (revefenacin) inhalation solution is an anticholinergic 2% and more common than placebo) include cough, indicated for the maintenance treatment of patients with chronic nasopharyngitis, upper respiratory tract infection, headache, and obstructive pulmonary disease (COPD). back pain. (6.1) -----------------DOSAGE AND ADMINISTRATION--------------- To report SUSPECTED ADVERSE REACTIONS, contact For oral inhalation use only. Do not swallow YUPELRI. Mylan at 1-877-446-3679 (1-877-4-INFO-RX) or FDA at 1-800 FDA-1088 or www.fda.gov/medwatch. • One 175 mcg vial (3 mL) once daily. (2) -----------------------DRUG INTERACTIONS------------------------ • For use with a standard jet nebulizer with a mouthpiece connected to an air compressor. (2) • Anticholinergics: May interact additively with concomitantly used anticholinergic medications. Avoid administration of ---------------DOSAGE FORMS AND STRENGTHS-------------- YUPELRI with other anticholinergic-containing drugs. (7.1) Inhalation solution in a unit-dose vial for nebulization. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data.