Genomic Platforms and Molecular Physiology of Insect Stress Tolerance
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diptera: Calyptratae)
Systematic Entomology (2020), DOI: 10.1111/syen.12443 Protein-encoding ultraconserved elements provide a new phylogenomic perspective of Oestroidea flies (Diptera: Calyptratae) ELIANA BUENAVENTURA1,2 , MICHAEL W. LLOYD2,3,JUAN MANUEL PERILLALÓPEZ4, VANESSA L. GONZÁLEZ2, ARIANNA THOMAS-CABIANCA5 andTORSTEN DIKOW2 1Museum für Naturkunde, Leibniz Institute for Evolution and Biodiversity Science, Berlin, Germany, 2National Museum of Natural History, Smithsonian Institution, Washington, DC, U.S.A., 3The Jackson Laboratory, Bar Harbor, ME, U.S.A., 4Department of Biological Sciences, Wright State University, Dayton, OH, U.S.A. and 5Department of Environmental Science and Natural Resources, University of Alicante, Alicante, Spain Abstract. The diverse superfamily Oestroidea with more than 15 000 known species includes among others blow flies, flesh flies, bot flies and the diverse tachinid flies. Oestroidea exhibit strikingly divergent morphological and ecological traits, but even with a variety of data sources and inferences there is no consensus on the relationships among major Oestroidea lineages. Phylogenomic inferences derived from targeted enrichment of ultraconserved elements or UCEs have emerged as a promising method for resolving difficult phylogenetic problems at varying timescales. To reconstruct phylogenetic relationships among families of Oestroidea, we obtained UCE loci exclusively derived from the transcribed portion of the genome, making them suitable for larger and more integrative phylogenomic studies using other genomic and transcriptomic resources. We analysed datasets containing 37–2077 UCE loci from 98 representatives of all oestroid families (except Ulurumyiidae and Mystacinobiidae) and seven calyptrate outgroups, with a total concatenated aligned length between 10 and 550 Mb. About 35% of the sampled taxa consisted of museum specimens (2–92 years old), of which 85% resulted in successful UCE enrichment. -

Arctic Expedition12° 16° 20° 24° 28° 32° Spitsbergen U Svalbard Archipelago 80° 80°
distinguished travel for more than 35 years Voyage UNDER THE Midnight Sun Arctic Expedition12° 16° 20° 24° 28° 32° Spitsbergen u Svalbard Archipelago 80° 80° 80° Raudfjorden Nordaustlandet Woodfjorden Smeerenburg Monaco Glacier The Arctic’s 79° 79° 79° Kongsfjorden Svalbard King’s Glacier Archipelago Ny-Ålesund Spitsbergen Longyearbyen Canada 78° 78° 78° i Greenland tic C rcle rc Sea Camp Millar A U.S. North Pole Russia Bellsund Calypsobyen Svalbard Archipelago Norway Copenhagen Burgerbukta 77° 77° 77° Cruise Itinerary Denmark Air Routing Samarin Glacier Hornsund Barents Sea June 20 to 30, 2022 4° 8° Spitsbergen12° u Samarin16° Glacier20° u Calypsobyen24° 76° 28° 32° 36° 76° Voyage across the Arctic Circle on this unique 11-day Monaco Glacier u Smeerenburg u Ny-Ålesund itinerary featuring a seven-night cruise round trip Copenhagen 1 Depart the U.S. or Canada aboard the Five-Star Le Boréal. Visit during the most 2 Arrive in Copenhagen, Denmark enchanting season, when the region is bathed in the magical 3 Copenhagen/Fly to Longyearbyen, Spitsbergen, light of the Midnight Sun. Cruise the shores of secluded Norway’s Svalbard Archipelago/Embark Le Boréal 4 Hornsund for Burgerbukta/Samarin Glacier Spitsbergen—the jewel of Norway’s rarely visited Svalbard 5 Bellsund for Calypsobyen/Camp Millar archipelago enjoy expert-led Zodiac excursions through 6 Cruising the Arctic Ice Pack sandstone mountain ranges, verdant tundra and awe-inspiring 7 MåkeØyane/Woodfjorden/Monaco Glacier ice formations. See glaciers calve in luminous blues and search 8 Raudfjorden for Smeerenburg for Arctic wildlife, including the “King of the Arctic,” the 9 Ny-Ålesund/Kongsfjorden for King’s Glacier polar bear, whales, walruses and Svalbard reindeer. -

Genomanalyse Von Prodiamesa Olivacea
Genomanalyse von Prodiamesa olivacea Dissertation zur Erlangung des Grades Doktor der Naturwissenschaften (Dr. rer. nat.) am Fachbereich Biologie der Johannes Gutenberg-Universität in Mainz Sarah Brunck geb. 08.08.1987 in Mainz Mainz, 2016 Dekan: 1. Berichterstatter: 2. Berichterstatter: Tag der mündlichen Prüfung: ii Inhaltsverzeichnis Inhaltsverzeichnis ................................................................................................................................ iii 1 Einleitung ........................................................................................................................................... 1 1.1 Die Familie der Chironomiden ................................................................................................. 1 1.1.1 Die Gattung Chironomus ..................................................................................................... 3 1.1.2 Die Gattung Prodiamesa ....................................................................................................... 6 1.2 Die Struktur von Insekten-Genomen am Beispiel der Chironomiden ............................... 9 1.2.1 Hochrepetitive DNA-Sequenzen ..................................................................................... 11 1.2.2 Mittelrepetitive DNA-Sequenzen bzw. Gen-Familien ................................................. 13 1.2.3 Gene und genregulatorische Sequenzen ........................................................................ 17 1.3 Zielsetzung ............................................................................................................................... -

Role of Epoxide Hydrolases in Lipid Metabolism
Biochimie 95 (2013) 91e95 Contents lists available at SciVerse ScienceDirect Biochimie journal homepage: www.elsevier.com/locate/biochi Mini-review Role of epoxide hydrolases in lipid metabolism Christophe Morisseau* Department of Entomology and U.C.D. Comprehensive Cancer Center, One Shields Avenue, University of California, Davis, CA 95616, USA article info abstract Article history: Epoxide hydrolases (EH), enzymes present in all living organisms, transform epoxide-containing lipids to Received 29 March 2012 1,2-diols by the addition of a molecule of water. Many of these oxygenated lipid substrates have potent Accepted 8 June 2012 biological activities: host defense, control of development, regulation of blood pressure, inflammation, Available online 18 June 2012 and pain. In general, the bioactivity of these natural epoxides is significantly reduced upon metabolism to diols. Thus, through the regulation of the titer of lipid epoxides, EHs have important and diverse bio- Keywords: logical roles with profound effects on the physiological state of the host organism. This review will Epoxide hydrolase discuss the biological activity of key lipid epoxides in mammals. In addition, the use of EH specific Epoxy-fatty acids Cholesterol epoxide inhibitors will be highlighted as possible therapeutic disease interventions. Ó Juvenile hormone 2012 Elsevier Masson SAS. All rights reserved. 1. Introduction hydrolyzed by a water molecule [8]. Based on this mechanism, transition-state inhibitors of EHs have been designed (Fig. 1B). Epoxides are three atom cyclic ethers formed by the oxidation of These ureas and amides are tight-binding competitive inhibitors olefins. Because of their highly polarized oxygen-carbon bonds and with low nanomolar dissociation constants (KI) [9] [10]. -

Eretmoptera Murphyi Schaeffer (Diptera: Chironomjdae), an Apparently Parthenogenetic Antarctic Midge
ERETMOPTERA MURPHYI SCHAEFFER (DIPTERA: CHIRONOMJDAE), AN APPARENTLY PARTHENOGENETIC ANTARCTIC MIDGE P. S. CRANSTON Entomology Department, British Museum (Natural History), Cromwell Road, London SWl SBD ABSTRACT. Chironomid midges are amongst the most abundant and diverse holo metabolous insects of the Antarctic and sub-Antarctic. Eretmoptera murphyi Schaeffer, 1914, has been enigmatic to systematists since the first discovery of adult females on South Georgia. The rediscovery of the species as a suspected introduction to Signy Island (South Orkney Islands) allows the description of the immature stages for the first time and the redescription of the female, the only sex known. E. murphyi larvae are terrestrial, living in damp moss and peat, and the brachypterous adult is probably parthenogenetic. Eretmoptera appears to have an isolated position amongst the terrestrial Orthocladiinae: the close relationship with the marine Clunio group of genera suggested by previous workers is not supported. INTRODUCTION In the Antarctic and sub-Antarctic regions, the Chironomidae (non-biting midges) are the commonest, most diverse and most widely distributed group ofhoi ometa bolo us insects. For example, Belgica antarctica Jacobs is the most southerly distributed free-living insect (Wirth and Gressitt, 1967; Usher and Edwards, 1984) and the podonomine genus Parochlus is found throughout the sub-Antarctic islands. Recently, Sublette and Wirth (1980) reported 22 species in 18 genera belonging to 6 subfamilies of Chironomidae from New Zealand's sub-Antarctic islands. One sub-Antarctic midge that has remained rather enigmatic since its discovery is Eretmoptera murphyi. Two females of this brachypterous chironomid were collected by R. C. Murphy from South Georgia in 1913 and described, together with other insects, by Schaeffer (1914). -

Continuous Up-Regulation of Heat Shock Proteins in Larvae, but Not Adults, of a Polar Insect
Continuous up-regulation of heat shock proteins in larvae, but not adults, of a polar insect Joseph P. Rinehart*†, Scott A. L. Hayward†‡, Michael A. Elnitsky§, Luke H. Sandro§, Richard E. Lee, Jr.§, and David L. Denlinger†¶ *Red River Valley Station, Agricultural Research Service, U.S. Department of Agriculture, Fargo, ND 58105; ‡School of Biological Sciences, Liverpool University, Liverpool L69 7ZB, United Kingdom; §Department of Zoology, Miami University, Oxford, OH 45056; and †Department of Entomology, Ohio State University, Columbus, OH 43210 Contributed by David L. Denlinger, August 8, 2006 Antarctica’s terrestrial environment is a challenge to which very the cessation of synthesis of most other proteins. Yet there are few animals have adapted. The largest, free-living animal to suggestions that some Antarctic species may respond somewhat inhabit the continent year-round is a flightless midge, Belgica differently. Both the Antarctic fish Trematomus bernacchii (5) antarctica. Larval midges survive the lengthy austral winter en- and an Antarctic ciliate, Euplotes focardii (6), constitutively cased in ice, and when the ice melts in summer, the larvae complete express hsp70 and show no or modest up-regulation of this gene their 2-yr life cycle, and the wingless adults form mating aggre- in response to thermal stress. This response is thought to be an gations while subjected to surprisingly high substrate tempera- adaptation to the cold, but constant, low temperature of the tures. Here we report a dichotomy in survival strategies exploited polar sea. The range of temperatures experienced in Antarctic by this insect at different stages of its life cycle. Larvae constitu- terrestrial environments, however, is quite different from that of tively up-regulate their heat shock proteins (small hsp, hsp70, and the ocean, and the midge faces a much broader range of hsp90) and maintain a high inherent tolerance to temperature temperatures in its natural habitat. -

Chironomidae Hirschkopf
Literatur Chironomidae Gesäuse U.A. zur Bestimmung und Ermittlung der Autökologie herangezogene Literatur: Albu, P. (1972): Două specii de Chironomide noi pentru ştiinţă în masivul Retezat.- St. şi Cerc. Biol., Seria Zoologie, 24: 15-20. Andersen, T.; Mendes, H.F. (2002): Neotropical and Mexican Mesosmittia Brundin, with the description of four new species (Insecta, Diptera, Chironomidae).- Spixiana, 25(2): 141-155. Andersen, T.; Sæther, O.A. (1993): Lerheimia, a new genus of Orthocladiinae from Africa (Diptera: Chironomidae).- Spixiana, 16: 105-112. Andersen, T.; Sæther, O.A.; Mendes, H.F. (2010): Neotropical Allocladius Kieffer, 1913 and Pseudosmittia Edwards, 1932 (Diptera: Chironomidae).- Zootaxa, 2472: 1-77. Baranov, V.A. (2011): New and rare species of Orthocladiinae (Diptera, Chironomidae) from the Crimea, Ukraine.- Vestnik zoologii, 45(5): 405-410. Boggero, A.; Zaupa, S.; Rossaro, B. (2014): Pseudosmittia fabioi sp. n., a new species from Sardinia (Diptera: Chironomidae, Orthocladiinae).- Journal of Entomological and Acarological Research, [S.l.],46(1): 1-5. Brundin, L. (1947): Zur Kenntnis der schwedischen Chironomiden.- Arkiv för Zoologi, 39 A(3): 1- 95. Brundin, L. (1956): Zur Systematik der Orthocladiinae (Dipt. Chironomidae).- Rep. Inst. Freshwat. Drottningholm 37: 5-185. Casas, J.J.; Laville, H. (1990): Micropsectra seguyi, n. sp. du groupe attenuata Reiss (Diptera: Chironomidae) de la Sierra Nevada (Espagne).- Annls Soc. ent. Fr. (N.S.), 26(3): 421-425. Caspers, N. (1983): Chironomiden-Emergenz zweier Lunzer Bäche, 1972.- Arch. Hydrobiol. Suppl. 65: 484-549. Caspers, N. (1987): Chaetocladius insolitus sp. n. (Diptera: Chironomidae) from Lunz, Austria. In: Saether, O.A. (Ed.): A conspectus of contemporary studies in Chironomidae (Diptera). -
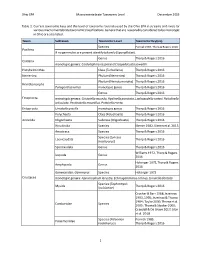
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 1 Table 1. Current Taxonomic Keys and the Level of Taxonomy Routinely U
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Table 1. Current taxonomic keys and the level of taxonomy routinely used by the Ohio EPA in streams and rivers for various macroinvertebrate taxonomic classifications. Genera that are reasonably considered to be monotypic in Ohio are also listed. Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Species Pennak 1989, Thorp & Rogers 2016 Porifera If no gemmules are present identify to family (Spongillidae). Genus Thorp & Rogers 2016 Cnidaria monotypic genera: Cordylophora caspia and Craspedacusta sowerbii Platyhelminthes Class (Turbellaria) Thorp & Rogers 2016 Nemertea Phylum (Nemertea) Thorp & Rogers 2016 Phylum (Nematomorpha) Thorp & Rogers 2016 Nematomorpha Paragordius varius monotypic genus Thorp & Rogers 2016 Genus Thorp & Rogers 2016 Ectoprocta monotypic genera: Cristatella mucedo, Hyalinella punctata, Lophopodella carteri, Paludicella articulata, Pectinatella magnifica, Pottsiella erecta Entoprocta Urnatella gracilis monotypic genus Thorp & Rogers 2016 Polychaeta Class (Polychaeta) Thorp & Rogers 2016 Annelida Oligochaeta Subclass (Oligochaeta) Thorp & Rogers 2016 Hirudinida Species Klemm 1982, Klemm et al. 2015 Anostraca Species Thorp & Rogers 2016 Species (Lynceus Laevicaudata Thorp & Rogers 2016 brachyurus) Spinicaudata Genus Thorp & Rogers 2016 Williams 1972, Thorp & Rogers Isopoda Genus 2016 Holsinger 1972, Thorp & Rogers Amphipoda Genus 2016 Gammaridae: Gammarus Species Holsinger 1972 Crustacea monotypic genera: Apocorophium lacustre, Echinogammarus ischnus, Synurella dentata Species (Taphromysis Mysida Thorp & Rogers 2016 louisianae) Crocker & Barr 1968; Jezerinac 1993, 1995; Jezerinac & Thoma 1984; Taylor 2000; Thoma et al. Cambaridae Species 2005; Thoma & Stocker 2009; Crandall & De Grave 2017; Glon et al. 2018 Species (Palaemon Pennak 1989, Palaemonidae kadiakensis) Thorp & Rogers 2016 1 Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Informal grouping of the Arachnida Hydrachnidia Smith 2001 water mites Genus Morse et al. -

Volume 2, Chapter 12-19: Terrestrial Insects: Holometabola-Diptera
Glime, J. M. 2017. Terrestrial Insects: Holometabola – Diptera Nematocera 2. In: Glime, J. M. Bryophyte Ecology. Volume 2. 12-19-1 Interactions. Ebook sponsored by Michigan Technological University and the International Association of Bryologists. eBook last updated 19 July 2020 and available at <http://digitalcommons.mtu.edu/bryophyte-ecology2/>. CHAPTER 12-19 TERRESTRIAL INSECTS: HOLOMETABOLA – DIPTERA NEMATOCERA 2 TABLE OF CONTENTS Cecidomyiidae – Gall Midges ........................................................................................................................ 12-19-2 Mycetophilidae – Fungus Gnats ..................................................................................................................... 12-19-3 Sciaridae – Dark-winged Fungus Gnats ......................................................................................................... 12-19-4 Ceratopogonidae – Biting Midges .................................................................................................................. 12-19-6 Chironomidae – Midges ................................................................................................................................. 12-19-9 Belgica .................................................................................................................................................. 12-19-14 Culicidae – Mosquitoes ................................................................................................................................ 12-19-15 Simuliidae – Blackflies -

Arctic Saga: Exploring Spitsbergen Via the Faroes and Jan Mayen Three
SPITSBERGEN, THE FAROES & JAN MAYEN SPITSBERGEN, GREENLAND & ICELAND Exploring the Fair Isle coastline; an encounter with Svalbard Reindeer; capturing the scenery. A Zodiac cruise with a view. Arctic Ocean GREENLAND SVALBARD Longyearbyen Bellsund Longyearbyen Hornsund Greenland Sea Arctic Saga: Barents GREENLAND Sea Three Arctic Islands: SVALBARD Jan Mayen FROM Exploring Spitsbergen via OSLO Norwegian Sea Spitsbergen, Greenland and Iceland SOUTHBOUND the Faroes and Jan Mayen Scoresby Atlantic Ocean A Sund RCT TO IC C OSLO Departing from Aberdeen, Scotland, the Arctic Saga voyage visits four remote IRCL Named one of the 50 Tours of a Lifetime by National Geographic Traveler, E Milne Ittoqqortoormiit Arctic islands. Sail through the North Atlantic to Fair Isle, famous for its bird Faroe Islands NORWAY this voyage offers the best of the eastern Arctic in one voyage. You start in Land observatory, followed by two days exploring the Viking and Norse sites on Shetland Spitsbergen, Norway, then sail south to Greenland to explore the world’s Denmark Islands Strait the Faroe Islands. Then it’s onto the world’s northernmost volcanic island, Atlantic Ocean Oslo largest fjord system and end in Iceland. There’s something for everyone: CLE Orkney C CIR ARCTI Jan Mayen. The last two days of your 14-day voyage are spent exploring Islands Fair Isle polar bears, walrus, muskoxen, local culture, ancient Thule settlements, Aberdeen Spitsbergen, always on the lookout for polar bears. hikes along the glacial moraine and tundra, and more. Reykjavik ICELAND Nature is the tour guide: Sea, ice, and weather conditions will determine your trip itinerary. Embrace the unexpected. -

ARTHROPODA Subphylum Hexapoda Protura, Springtails, Diplura, and Insects
NINE Phylum ARTHROPODA SUBPHYLUM HEXAPODA Protura, springtails, Diplura, and insects ROD P. MACFARLANE, PETER A. MADDISON, IAN G. ANDREW, JOCELYN A. BERRY, PETER M. JOHNS, ROBERT J. B. HOARE, MARIE-CLAUDE LARIVIÈRE, PENELOPE GREENSLADE, ROSA C. HENDERSON, COURTenaY N. SMITHERS, RicarDO L. PALMA, JOHN B. WARD, ROBERT L. C. PILGRIM, DaVID R. TOWNS, IAN McLELLAN, DAVID A. J. TEULON, TERRY R. HITCHINGS, VICTOR F. EASTOP, NICHOLAS A. MARTIN, MURRAY J. FLETCHER, MARLON A. W. STUFKENS, PAMELA J. DALE, Daniel BURCKHARDT, THOMAS R. BUCKLEY, STEVEN A. TREWICK defining feature of the Hexapoda, as the name suggests, is six legs. Also, the body comprises a head, thorax, and abdomen. The number A of abdominal segments varies, however; there are only six in the Collembola (springtails), 9–12 in the Protura, and 10 in the Diplura, whereas in all other hexapods there are strictly 11. Insects are now regarded as comprising only those hexapods with 11 abdominal segments. Whereas crustaceans are the dominant group of arthropods in the sea, hexapods prevail on land, in numbers and biomass. Altogether, the Hexapoda constitutes the most diverse group of animals – the estimated number of described species worldwide is just over 900,000, with the beetles (order Coleoptera) comprising more than a third of these. Today, the Hexapoda is considered to contain four classes – the Insecta, and the Protura, Collembola, and Diplura. The latter three classes were formerly allied with the insect orders Archaeognatha (jumping bristletails) and Thysanura (silverfish) as the insect subclass Apterygota (‘wingless’). The Apterygota is now regarded as an artificial assemblage (Bitsch & Bitsch 2000). -

Disruption of Soluble Epoxide Hydrolase Dimerization As a Novel Therapeutic Target for Stroke
DISRUPTION OF SOLUBLE EPOXIDE HYDROLASE DIMERIZATION AS A NOVEL THERAPEUTIC TARGET FOR STROKE By Jonathan W. Nelson A THESIS/DISSERTATION Presented to the Department of Molecular and Medical Genetics and the Oregon Health & Science University School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy April 2013 TABLE OF CONTENTS List of Figures ...................................................................................................................... vi List of Tables ...................................................................................................................... vii List of Abbreviations ......................................................................................................... viii Acknowledgements ........................................................................................................... xiii Dissertation Abstract ........................................................................................................ xvi Chapter 1 Targeting Soluble Epoxide Hydrolase Dimerization As a Stroke Therapy ......... 1 Abstract ........................................................................................................................... 1 Stroke and Society ........................................................................................................... 2 Prevention and Treatment of Stroke .............................................................................. 3 Soluble Epoxide Hydrolase and Stroke ..........................................................................