SCIENCE CHINA Complete Chloroplast Genome Sequence Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
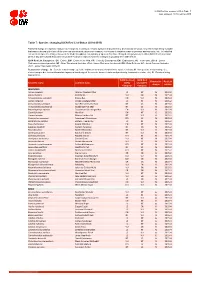
Table 7: Species Changing IUCN Red List Status (2014-2015)
IUCN Red List version 2015.4: Table 7 Last Updated: 19 November 2015 Table 7: Species changing IUCN Red List Status (2014-2015) Published listings of a species' status may change for a variety of reasons (genuine improvement or deterioration in status; new information being available that was not known at the time of the previous assessment; taxonomic changes; corrections to mistakes made in previous assessments, etc. To help Red List users interpret the changes between the Red List updates, a summary of species that have changed category between 2014 (IUCN Red List version 2014.3) and 2015 (IUCN Red List version 2015-4) and the reasons for these changes is provided in the table below. IUCN Red List Categories: EX - Extinct, EW - Extinct in the Wild, CR - Critically Endangered, EN - Endangered, VU - Vulnerable, LR/cd - Lower Risk/conservation dependent, NT - Near Threatened (includes LR/nt - Lower Risk/near threatened), DD - Data Deficient, LC - Least Concern (includes LR/lc - Lower Risk, least concern). Reasons for change: G - Genuine status change (genuine improvement or deterioration in the species' status); N - Non-genuine status change (i.e., status changes due to new information, improved knowledge of the criteria, incorrect data used previously, taxonomic revision, etc.); E - Previous listing was an Error. IUCN Red List IUCN Red Reason for Red List Scientific name Common name (2014) List (2015) change version Category Category MAMMALS Aonyx capensis African Clawless Otter LC NT N 2015-2 Ailurus fulgens Red Panda VU EN N 2015-4 -

Officinalis Var. Biloba and Eucommia Ulmoides in Traditional Chinese Medicine
Two Thousand Years of Eating Bark: Magnolia officinalis var. biloba and Eucommia ulmoides in Traditional Chinese Medicine Todd Forrest With a sense of urgency inspired by the rapid disappearance of plant habitats, most researchers are focusing on tropical flora as the source of plant-based medicines. However, new medicines may also be developed from plants of the world’s temperate regions. While working in his garden in the spring of English yew (Taxus baccata), a species common 1763, English clergyman Edward Stone was in cultivation. Foxglove (Digitalis purpurea), positive he had found a cure for malaria. Tasting the source of digitoxin, had a long history as the bark of a willow (Salix alba), Stone noticed a folk medicine in England before 1775, when a bitter flavor similar to that of fever tree (Cin- William Withering found it to be an effective chona spp.), the Peruvian plant used to make cure for dropsy. Doctors now prescribe digitoxin quinine. He reported his discovery to the Royal as a treatment for congestive heart failure. Society in London, recommending that willow EGb 761, a compound extracted from the be tested as an inexpensive alternative to fever maidenhair tree (Gingko biloba), is another ex- tree. Although experiments revealed that wil- ample of a drug developed from a plant native to low bark could not cure malaria, it did reduce the North Temperate Zone. Used as an herbal some of the feverish symptoms of the disease. remedy in China for centuries, ginkgo extract is Based on these findings, Stone’s simple taste now packaged and marketed in the West as a test led to the development of a drug used every treatment for ailments ranging from short-term day around the world: willow bark was the first memory loss to impotence. -
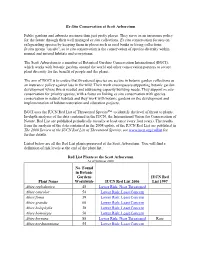
IUCN Red List of Threatened Species™ to Identify the Level of Threat to Plants
Ex-Situ Conservation at Scott Arboretum Public gardens and arboreta are more than just pretty places. They serve as an insurance policy for the future through their well managed ex situ collections. Ex situ conservation focuses on safeguarding species by keeping them in places such as seed banks or living collections. In situ means "on site", so in situ conservation is the conservation of species diversity within normal and natural habitats and ecosystems. The Scott Arboretum is a member of Botanical Gardens Conservation International (BGCI), which works with botanic gardens around the world and other conservation partners to secure plant diversity for the benefit of people and the planet. The aim of BGCI is to ensure that threatened species are secure in botanic garden collections as an insurance policy against loss in the wild. Their work encompasses supporting botanic garden development where this is needed and addressing capacity building needs. They support ex situ conservation for priority species, with a focus on linking ex situ conservation with species conservation in natural habitats and they work with botanic gardens on the development and implementation of habitat restoration and education projects. BGCI uses the IUCN Red List of Threatened Species™ to identify the level of threat to plants. In-depth analyses of the data contained in the IUCN, the International Union for Conservation of Nature, Red List are published periodically (usually at least once every four years). The results from the analysis of the data contained in the 2008 update of the IUCN Red List are published in The 2008 Review of the IUCN Red List of Threatened Species; see www.iucn.org/redlist for further details. -

Relora-Plex Supports a Healthy Stress Response†
PRODUCT DATA DOUGLAS LABORATORIES® 07/2015 1 Relora-Plex Supports a Healthy Stress Response† DESCRIPTION Relora-Plex is a unique proprietary blend of two herbal extracts, Magnolia and Phellodendron, combined with B vitamins designed to support normal mental functioning during stress. Relora® was shown in published clinical studies to support normal salivary cortisol levels, stress, mood, and weight management.† FUNCTIONS Cortisol, a hormone produced in the adrenal glands, plays an important role in the body’s regulation of cardiovascular function and fat, protein and carbohydrate utilization. When the body experiences stress, cortisol secretion increases, thus causing a breakdown of muscle protein and the release of amino acids to form glucose via gluconeogenesis. The resulting higher level of glucose in the body, combined with the decreased use of glucose by other tissues in the body, ensures that the brain is receiving adequate energy. Continuing research indicates that stress and anxiety can have a significant impact on the body’s health and wellbeing. While cortisol secretion is an important part of the body’s response to stress, the prolonged secretion of cortisol can have detrimental effects to the proper functioning of the body’s cardiovascular, immune, neurological and metabolic systems. Relora® is a patent-pending combination of two herbal extracts of Magnolia and Phellodendron bark (Asian cork tree). Both herbs have been used in Traditional Chinese Medicine for several hundred years. In a human study, 82% of the participants taking Relora® agreed with the statement that: “Relora® helps control…. irritability, emotional ups and downs, restlessness, tense muscles, poor sleep, fatigue, and concentration difficulties.”* † Relora® was found not to cause sedation, though 74% of the patients had more restful sleep. -

Therapeutic Applications of Compounds in the Magnolia Family
Pharmacology & Therapeutics 130 (2011) 157–176 Contents lists available at ScienceDirect Pharmacology & Therapeutics journal homepage: www.elsevier.com/locate/pharmthera Associate Editor: I. Kimura Therapeutic applications of compounds in the Magnolia family Young-Jung Lee a, Yoot Mo Lee a,b, Chong-Kil Lee a, Jae Kyung Jung a, Sang Bae Han a, Jin Tae Hong a,⁎ a College of Pharmacy and Medical Research Center, Chungbuk National University, 12 Gaesin-dong, Heungduk-gu, Cheongju, Chungbuk 361-763, Republic of Korea b Reviewer & Scientificofficer, Bioequivalence Evaluation Division, Drug Evaluation Department Pharmaceutical Safety Breau, Korea Food & Drug Administration, Republic of Korea article info abstract Keywords: The bark and/or seed cones of the Magnolia tree have been used in traditional herbal medicines in Korea, Magnolia China and Japan. Bioactive ingredients such as magnolol, honokiol, 4-O-methylhonokiol and obovatol have Magnolol received great attention, judging by the large number of investigators who have studied their Obovatol pharmacological effects for the treatment of various diseases. Recently, many investigators reported the Honokiol anti-cancer, anti-stress, anti-anxiety, anti-depressant, anti-oxidant, anti-inflammatory and hepatoprotective 4-O-methylhonokiol effects as well as toxicities and pharmacokinetics data, however, the mechanisms underlying these Cancer Nerve pharmacological activities are not clear. The aim of this study was to review a variety of experimental and Alzheimer disease clinical reports and, describe the effectiveness, toxicities and pharmacokinetics, and possible mechanisms of Cardiovascular disease Magnolia and/or its constituents. Inflammatory disease © 2011 Elsevier Inc. All rights reserved. Contents 1. Introduction .............................................. 157 2. Components of Magnolia ........................................ 159 3. Therapeutic applications in cancer ................................... -

Magnolia Obovata
ISSUE 80 INAGNOLN INagnolla obovata Eric Hsu, Putnam Fellow, Arnold Arboretum of Harvard University Photographs by Philippe de 8 poelberch I first encountered Magnolia obovata in Bower at Sir Harold Hillier Gardens and Arboretum, Hampshire, England, where the tightly pursed, waxy, globular buds teased, but rewarded my patience. As each bud unfurled successively, it emitted an intoxicating ambrosial bouquet of melons, bananas, and grapes. Although the leaves were nowhere as luxuriously lustrous as M. grandrflora, they formed an el- egant wreath for the creamy white flower. I gingerly plucked one flower for doser observation, and placed one in my room. When I re- tumed from work later in the afternoon, the mom was overpowering- ly redolent of the magnolia's scent. The same olfactory pleasure was later experienced vicariously through the large Magnolia x wiesneri in the private garden of Nicholas Nickou in southern Connecticut. Several years earlier, I had traveled to Hokkaido Japan, after my high school graduation. Although Hokkaido experiences more severe win- ters than those in the southern parts of Japan, the forests there yield a remarkable diversity of fora, some of which are popular ornamen- tals. When one drives through the region, the silvery to blue-green leaf undersides of Magnolia obovata, shimmering in the breeze, seem to flag the eyes. In "Forest Flora of Japan" (sggII), Charles Sargent commended this species, which he encountered growing tluough the mountainous forests of Hokkaido. He called it "one of the largest and most beautiful of the deciduous-leaved species in size and [the seed conesj are sometimes eight inches long, and brilliant scarlet in color, stand out on branches, it is the most striking feature of the forests. -

The Progressive and Ancestral Traits of the Secondary Xylem Within Magnolia Clad – the Early Diverging Lineage of Flowering Plants
Acta Societatis Botanicorum Poloniae ORIGINAL RESEARCH PAPER Acta Soc Bot Pol 84(1):87–96 DOI: 10.5586/asbp.2014.028 Received: 2014-07-31 Accepted: 2014-12-01 Published electronically: 2015-01-07 The progressive and ancestral traits of the secondary xylem within Magnolia clad – the early diverging lineage of flowering plants Magdalena Marta Wróblewska* Department of Developmental Plant Biology, Institute of Experimental Biology, University of Wrocław, Kanonia 6/8, 50-328 Wrocław, Poland Abstract The qualitative and quantitative studies, presented in this article, on wood anatomy of various species belonging to ancient Magnolia genus reveal new aspects of phylogenetic relationships between the species and show evolutionary trends, known to increase fitness of conductive tissues in angiosperms. They also provide new examples of phenotypic plasticity in plants. The type of perforation plate in vessel members is one of the most relevant features for taxonomic studies. InMagnolia , until now, two types of perforation plates have been reported: the conservative, scalariform and the specialized, simple one. In this paper, are presented some findings, new to magnolia wood science, like exclusively simple perforation plates in some species or mixed perforation plates – simple and scalariform in one vessel member. Intravascular pitting is another taxonomically important trait of vascular tissue. Interesting transient states between different patterns of pitting in one cell only have been found. This proves great flexibility of mechanisms, which elaborate cell wall structure in maturing trache- ary element. The comparison of this data with phylogenetic trees, based on the fossil records and plastid gene expression, clearly shows that there is a link between the type of perforation plate and the degree of evolutionary specialization within Magnolia genus. -

WO 2010/002406 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 7 January 2010 (07.01.2010) WO 2010/002406 Al (51) International Patent Classification: EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, A61K 36/00 (2006.01) HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, PCT/US2008/069017 NZ, OM, PG, PH, PL, PT, RO, RS, RU, SC, SD, SE, SG, (22) International Filing Date: SK, SL, SM, SV, SY, TJ, TM, TN, TR, TT, TZ, UA, UG, 2 July 2008 (02.07.2008) US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, (71) Applicant and ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, (72) Inventor: JAFFE, Russell, M. [US/US]; 10430 Hunter TM), European (AT, BE, BG, CH, CY, CZ, DE, DK, EE, View Road, Vienna, VA 22181 (US). ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, MC, MT, NL, NO, PL, PT, RO, SE, SI, SK, TR), OAPI (74) Agents: WARREN, Leigh, M. et al; Cooley Godward (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, ML, MR, Kronish LLP, 777 6th Street, N.W., Suite 1100, Washing NE, SN, TD, TG). -

Magnolia Officinalis Rehder & Wilson
Magnolia Officinalis Rehder & wilson This fine Magnolia is closely related when it is possible to compare living to, and has been confused with, the flowers, one wtth another, other Japanese M. obvata, better known in differences will be noted. The different gardens as M. hvpoleuca and naturally color of the branches is an infallible enough since the foliage of the two character by which to distmguish the species is identical. There are, however, two species at any season of the year. in bark and fruit many noticeable On the mountains of western Hupeh differences. The Japanese species has a and Szechuan up to elevations of 6000 purplish bark on shoots one year old. feet above sea level M. officinalis is the staminal and carpellary column is commonly cultivated, but I never met about two inches long, acutish at the with a spontaneous tree in the forests. summit, and the filaments are more This remark would apply to many than two thirds as long as the anthers. other Chinese trees of economic value The fruit is cylindric. stx to eight (Gleditsia officinalis and Eucommia inches long, about two inches wide, ulmoides, for example) but there is no somewhat pointed at the apex and reason to doubt that all of them are attenuate at the base. The ripe carpels truly natives of the region where they have long, usually slightly recurved are also found cultivated. Trees valued beaks, and rather thin walls. Doubtless for the medicinal value of their bark Rowers of M. officinalis (left) and M. fraseri (right) opened simulraneously on adjacent trees in Phil Seitner s garden. -

Magnolia Wilsonii Bill Mcnamara, Executive Director Quarrytu'll Botanic Garden
Nagnolia Issue 88 Endangered: Magnolia wilsonii Bill McNamara, Executive Director Quarrytu'll Botanic Garden My choice for a favorite tree seems to change almost weekly, sometimes even daily. But one has currently held my impatient mind longer than normal. It is a tree with so many admirable qualities that I think everyone should try to find a place for it in their gardens. Though widely available in the nursery trade, it is also a tree that may soon be extinct in the wild. Imagine a tree that blooms after the danger of frosts, has pristine white 12-centimeter-wide flowers with brilliant crimson-red stamens that hang like bright lantern, has an elegant rich fragrance, doesn't get too big, and is named after the indefatigable plant hunter, E. H. Wilson. Of course, I'm thinking of Magnolia milsanii (Finet & Gagnep. ) Rehder, an endangered tree native to China occurring in western Sichuan, northern Yunnan and western Guizhou. Habitat loss and fragmentation have caused its num- bers in the wild to decrease to a dangerously low level. It is found in mixed forests between 1900 and 3300 meters elevation. Much of its habi- tat, once one of the richest temperate forests on earth, has been reduced to small areas too steep to clear for agriculture. Another threat to its sur- vival in the wild is demand for its bark due to its medicinal qualities. It is frequently used as a substitute for the bark of Magnolia officinalis. For centuries in China bark from several magnolia species has been used to clear head and chest congestion and for intestinal relief, while flower buds of many species have been used for sinus congestion. -

Decorative Magnolia Plants: a Comparison of the Content of Their Biologically Active Components Showing Antimicrobial Effects
plants Article Decorative Magnolia Plants: A Comparison of the Content of Their Biologically Active Components Showing Antimicrobial Effects Petra Lovecká 1,*, AlžbˇetaSvobodová 1, Anna Mac ˚urková 1, Blanka Vrchotová 1, KateˇrinaDemnerová 1 and ZdenˇekWimmer 2,3 1 Department of Biochemistry and Microbiology, University of Chemistry and Technology in Prague, Technická 3, 166 28 Prague 6, Czech Republic; [email protected] (A.S.); [email protected] (A.M.); [email protected] (B.V.); [email protected] (K.D.) 2 Department of Chemistry of Natural Compounds, University of Chemistry and Technology in Prague, Technická 5, 166 28 Prague 6, Czech Republic; [email protected] 3 Isotope Laboratory, Institute of Experimental Botany, Czech Academy of Sciences, Vídeˇnská 1083, 142 20 Prague 4, Czech Republic * Correspondence: [email protected]; Tel.: +420-220-445-139 Received: 19 May 2020; Accepted: 9 July 2020; Published: 11 July 2020 Abstract: Magnolia plants are used both as food supplements and as cosmetic and medicinal products. The objectives of this work consisted of preparing extracts from leaves and flowers of eight Magnolia plants, and of determining concentrations of magnolol (1 to 100 mg g 1), honokiol (0.11 to 250 mg g 1), · − · − and obovatol (0.09 to 650 mg g 1), typical neolignans for the genus Magnolia, in extracts made by using · − a methanol/water (80/20) mixture. The tested Magnolia plants, over sixty years old, were obtained from Pr ˚uhonický Park (Prague area, Czech Republic): M. tripetala MTR 1531, M. obovata MOB 1511, and six hybrid plants Magnolia pruhoniciana, results of a crossbreeding of M. -

Houpu Magnolia Magnolia Officinalis Var. Biloba
Hidden Treasures of the Arboretum Houpu Magnolia Magnolia officinalis var. biloba B Y D ANIEL M OUNT he Arboretum’s magnolia collection occurs. This rather scattered distribution is due to is a great joy to visitors each spring. the age of the genus, about 100 million years, and T About 250 specimens—the third- the fact it has survived many geological changes largest collection in the country—grace the park and upheavals. As for the plants themselves, they with their primordial beauty. Many of these are have changed little in their long tenure on Earth. cultivars, but there is also a nice representa- Among the many showy-flowered species, tion of species: Twenty-six of the 120 magnolia cultivars and hybrid magnolias in the Arboretum, species in the genus worldwide grow here. one is notable for the uniqueness of its foliage: Magnolias, considered to be among the first Magnolia officinalis, commonly known as the flowering plants, can be found growing in the Houpu or spice magnolia. A deciduous tree native wild throughout the Americas and Southeast to China, M. officinalis grows from 20 to 50 feet tall Asia, where the highest concentration of species and is found at elevations of 300 to 1500 meters 12 v Washington Park Arboretum Bulletin LEFT: Magnolia officinalis var. biloba blooming near the Hybrid Rhododendron Garden in early May. It’s prized for it beautiful flowers and large, notched leaves. (Photo by Niall Dunne) rounded leaf tip, while the latter has a deep notch on the tips of its leaves. Magnolia expert G.H. Johnstone observed that many M.