The Genome Assembly and Annotation of Magnolia Biondii Pamp., A
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
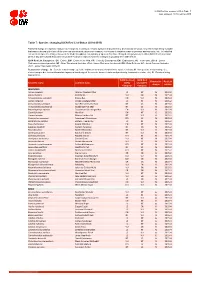
Table 7: Species Changing IUCN Red List Status (2014-2015)
IUCN Red List version 2015.4: Table 7 Last Updated: 19 November 2015 Table 7: Species changing IUCN Red List Status (2014-2015) Published listings of a species' status may change for a variety of reasons (genuine improvement or deterioration in status; new information being available that was not known at the time of the previous assessment; taxonomic changes; corrections to mistakes made in previous assessments, etc. To help Red List users interpret the changes between the Red List updates, a summary of species that have changed category between 2014 (IUCN Red List version 2014.3) and 2015 (IUCN Red List version 2015-4) and the reasons for these changes is provided in the table below. IUCN Red List Categories: EX - Extinct, EW - Extinct in the Wild, CR - Critically Endangered, EN - Endangered, VU - Vulnerable, LR/cd - Lower Risk/conservation dependent, NT - Near Threatened (includes LR/nt - Lower Risk/near threatened), DD - Data Deficient, LC - Least Concern (includes LR/lc - Lower Risk, least concern). Reasons for change: G - Genuine status change (genuine improvement or deterioration in the species' status); N - Non-genuine status change (i.e., status changes due to new information, improved knowledge of the criteria, incorrect data used previously, taxonomic revision, etc.); E - Previous listing was an Error. IUCN Red List IUCN Red Reason for Red List Scientific name Common name (2014) List (2015) change version Category Category MAMMALS Aonyx capensis African Clawless Otter LC NT N 2015-2 Ailurus fulgens Red Panda VU EN N 2015-4 -

Officinalis Var. Biloba and Eucommia Ulmoides in Traditional Chinese Medicine
Two Thousand Years of Eating Bark: Magnolia officinalis var. biloba and Eucommia ulmoides in Traditional Chinese Medicine Todd Forrest With a sense of urgency inspired by the rapid disappearance of plant habitats, most researchers are focusing on tropical flora as the source of plant-based medicines. However, new medicines may also be developed from plants of the world’s temperate regions. While working in his garden in the spring of English yew (Taxus baccata), a species common 1763, English clergyman Edward Stone was in cultivation. Foxglove (Digitalis purpurea), positive he had found a cure for malaria. Tasting the source of digitoxin, had a long history as the bark of a willow (Salix alba), Stone noticed a folk medicine in England before 1775, when a bitter flavor similar to that of fever tree (Cin- William Withering found it to be an effective chona spp.), the Peruvian plant used to make cure for dropsy. Doctors now prescribe digitoxin quinine. He reported his discovery to the Royal as a treatment for congestive heart failure. Society in London, recommending that willow EGb 761, a compound extracted from the be tested as an inexpensive alternative to fever maidenhair tree (Gingko biloba), is another ex- tree. Although experiments revealed that wil- ample of a drug developed from a plant native to low bark could not cure malaria, it did reduce the North Temperate Zone. Used as an herbal some of the feverish symptoms of the disease. remedy in China for centuries, ginkgo extract is Based on these findings, Stone’s simple taste now packaged and marketed in the West as a test led to the development of a drug used every treatment for ailments ranging from short-term day around the world: willow bark was the first memory loss to impotence. -

Magnolia Finally Flowers in Boston
The "Hope of Spring" Magnolia Finally Flowers in Boston Stephen A. Spongberg and Peter Del T~edici After a difficult start, Magnolia biondii from China flowered in the Arboretum for the first time in March of 1991. The spring and early summer of 1991 at the biloba, and Magnolia biondii. While we were Arnold Arboretum were extraordinary with eager to examine each of these in turn, and regard to the heavy flowering of many of the to document their flowering with voucher trees and shrubs within the Arboretum’s col- herbarium specimens and photographs, the lections. Nor was this phenomenon restricted first flowering of the last-named magnolia to the confines of the Arboretum, for across presented us with the opportunity to examine the Northeast crabapples, flv~y.riWg dog- the flowers of tills species and to fix its posi- woods, and other ornamental trees and shrubs tion in the classification of the genus T A _ _ _ _ _ 7 ’ »Ync~llrar~ an ~lwnr~~n~nr ~f 1-.lv~,-,.‘ ‘ly.y i ^..t_^.7 tvtu~ttVllCt. the season as outstanding. The relatively mild winter of 1990-1991 and the abundant rainfall Early History of the Species that fell during the summer of 1990 combined Magnolia biondii was first described by the to make the spring of 1991 an exceptionally Italian botanist Renato Pampanini in 1910 floriferous one. based on specimens collected in Hubei Not only was there an abundance of bloom, Province in central China in 1906 by the but many of the newer accessions at the Italian missionary and naturalist, P. -

THE Magnoliaceae Liriodendron L. Magnolia L
THE Magnoliaceae Liriodendron L. Magnolia L. VEGETATIVE KEY TO SPECIES IN CULTIVATION Jan De Langhe (1 October 2014 - 28 May 2015) Vegetative identification key. Introduction: This key is based on vegetative characteristics, and therefore also of use when flowers and fruits are absent. - Use a 10× hand lens to evaluate stipular scars, buds and pubescence in general. - Look at the entire plant. Young specimens, shade, and strong shoots give an atypical view. - Beware of hybridisation, especially with plants raised from seed other than wild origin. Taxa treated in this key: see page 10. Questionable/frequently misapplied names: see page 10. Names referred to synonymy: see page 11. References: - JDL herbarium - living specimens, in various arboreta, botanic gardens and collections - literature: De Meyere, D. - (2001) - Enkele notities omtrent Liriodendron tulipifera, L. chinense en hun hybriden in BDB, p.23-40. Hunt, D. - (1998) - Magnolias and their allies, 304p. Bean, W.J. - (1981) - Magnolia in Trees and Shrubs hardy in the British Isles VOL.2, p.641-675. - or online edition Clarke, D.L. - (1988) - Magnolia in Trees and Shrubs hardy in the British Isles supplement, p.318-332. Grimshaw, J. & Bayton, R. - (2009) - Magnolia in New Trees, p.473-506. RHS - (2014) - Magnolia in The Hillier Manual of Trees & Shrubs, p.206-215. Liu, Y.-H., Zeng, Q.-W., Zhou, R.-Z. & Xing, F.-W. - (2004) - Magnolias of China, 391p. Krüssmann, G. - (1977) - Magnolia in Handbuch der Laubgehölze, VOL.3, p.275-288. Meyer, F.G. - (1977) - Magnoliaceae in Flora of North America, VOL.3: online edition Rehder, A. - (1940) - Magnoliaceae in Manual of cultivated trees and shrubs hardy in North America, p.246-253. -
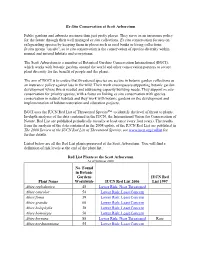
IUCN Red List of Threatened Species™ to Identify the Level of Threat to Plants
Ex-Situ Conservation at Scott Arboretum Public gardens and arboreta are more than just pretty places. They serve as an insurance policy for the future through their well managed ex situ collections. Ex situ conservation focuses on safeguarding species by keeping them in places such as seed banks or living collections. In situ means "on site", so in situ conservation is the conservation of species diversity within normal and natural habitats and ecosystems. The Scott Arboretum is a member of Botanical Gardens Conservation International (BGCI), which works with botanic gardens around the world and other conservation partners to secure plant diversity for the benefit of people and the planet. The aim of BGCI is to ensure that threatened species are secure in botanic garden collections as an insurance policy against loss in the wild. Their work encompasses supporting botanic garden development where this is needed and addressing capacity building needs. They support ex situ conservation for priority species, with a focus on linking ex situ conservation with species conservation in natural habitats and they work with botanic gardens on the development and implementation of habitat restoration and education projects. BGCI uses the IUCN Red List of Threatened Species™ to identify the level of threat to plants. In-depth analyses of the data contained in the IUCN, the International Union for Conservation of Nature, Red List are published periodically (usually at least once every four years). The results from the analysis of the data contained in the 2008 update of the IUCN Red List are published in The 2008 Review of the IUCN Red List of Threatened Species; see www.iucn.org/redlist for further details. -

Relora-Plex Supports a Healthy Stress Response†
PRODUCT DATA DOUGLAS LABORATORIES® 07/2015 1 Relora-Plex Supports a Healthy Stress Response† DESCRIPTION Relora-Plex is a unique proprietary blend of two herbal extracts, Magnolia and Phellodendron, combined with B vitamins designed to support normal mental functioning during stress. Relora® was shown in published clinical studies to support normal salivary cortisol levels, stress, mood, and weight management.† FUNCTIONS Cortisol, a hormone produced in the adrenal glands, plays an important role in the body’s regulation of cardiovascular function and fat, protein and carbohydrate utilization. When the body experiences stress, cortisol secretion increases, thus causing a breakdown of muscle protein and the release of amino acids to form glucose via gluconeogenesis. The resulting higher level of glucose in the body, combined with the decreased use of glucose by other tissues in the body, ensures that the brain is receiving adequate energy. Continuing research indicates that stress and anxiety can have a significant impact on the body’s health and wellbeing. While cortisol secretion is an important part of the body’s response to stress, the prolonged secretion of cortisol can have detrimental effects to the proper functioning of the body’s cardiovascular, immune, neurological and metabolic systems. Relora® is a patent-pending combination of two herbal extracts of Magnolia and Phellodendron bark (Asian cork tree). Both herbs have been used in Traditional Chinese Medicine for several hundred years. In a human study, 82% of the participants taking Relora® agreed with the statement that: “Relora® helps control…. irritability, emotional ups and downs, restlessness, tense muscles, poor sleep, fatigue, and concentration difficulties.”* † Relora® was found not to cause sedation, though 74% of the patients had more restful sleep. -

Therapeutic Applications of Compounds in the Magnolia Family
Pharmacology & Therapeutics 130 (2011) 157–176 Contents lists available at ScienceDirect Pharmacology & Therapeutics journal homepage: www.elsevier.com/locate/pharmthera Associate Editor: I. Kimura Therapeutic applications of compounds in the Magnolia family Young-Jung Lee a, Yoot Mo Lee a,b, Chong-Kil Lee a, Jae Kyung Jung a, Sang Bae Han a, Jin Tae Hong a,⁎ a College of Pharmacy and Medical Research Center, Chungbuk National University, 12 Gaesin-dong, Heungduk-gu, Cheongju, Chungbuk 361-763, Republic of Korea b Reviewer & Scientificofficer, Bioequivalence Evaluation Division, Drug Evaluation Department Pharmaceutical Safety Breau, Korea Food & Drug Administration, Republic of Korea article info abstract Keywords: The bark and/or seed cones of the Magnolia tree have been used in traditional herbal medicines in Korea, Magnolia China and Japan. Bioactive ingredients such as magnolol, honokiol, 4-O-methylhonokiol and obovatol have Magnolol received great attention, judging by the large number of investigators who have studied their Obovatol pharmacological effects for the treatment of various diseases. Recently, many investigators reported the Honokiol anti-cancer, anti-stress, anti-anxiety, anti-depressant, anti-oxidant, anti-inflammatory and hepatoprotective 4-O-methylhonokiol effects as well as toxicities and pharmacokinetics data, however, the mechanisms underlying these Cancer Nerve pharmacological activities are not clear. The aim of this study was to review a variety of experimental and Alzheimer disease clinical reports and, describe the effectiveness, toxicities and pharmacokinetics, and possible mechanisms of Cardiovascular disease Magnolia and/or its constituents. Inflammatory disease © 2011 Elsevier Inc. All rights reserved. Contents 1. Introduction .............................................. 157 2. Components of Magnolia ........................................ 159 3. Therapeutic applications in cancer ................................... -

Magnolia Obovata
ISSUE 80 INAGNOLN INagnolla obovata Eric Hsu, Putnam Fellow, Arnold Arboretum of Harvard University Photographs by Philippe de 8 poelberch I first encountered Magnolia obovata in Bower at Sir Harold Hillier Gardens and Arboretum, Hampshire, England, where the tightly pursed, waxy, globular buds teased, but rewarded my patience. As each bud unfurled successively, it emitted an intoxicating ambrosial bouquet of melons, bananas, and grapes. Although the leaves were nowhere as luxuriously lustrous as M. grandrflora, they formed an el- egant wreath for the creamy white flower. I gingerly plucked one flower for doser observation, and placed one in my room. When I re- tumed from work later in the afternoon, the mom was overpowering- ly redolent of the magnolia's scent. The same olfactory pleasure was later experienced vicariously through the large Magnolia x wiesneri in the private garden of Nicholas Nickou in southern Connecticut. Several years earlier, I had traveled to Hokkaido Japan, after my high school graduation. Although Hokkaido experiences more severe win- ters than those in the southern parts of Japan, the forests there yield a remarkable diversity of fora, some of which are popular ornamen- tals. When one drives through the region, the silvery to blue-green leaf undersides of Magnolia obovata, shimmering in the breeze, seem to flag the eyes. In "Forest Flora of Japan" (sggII), Charles Sargent commended this species, which he encountered growing tluough the mountainous forests of Hokkaido. He called it "one of the largest and most beautiful of the deciduous-leaved species in size and [the seed conesj are sometimes eight inches long, and brilliant scarlet in color, stand out on branches, it is the most striking feature of the forests. -

Fragrant Annuals Fragrant Annuals
TheThe AmericanAmerican GARDENERGARDENER® TheThe MagazineMagazine ofof thethe AAmericanmerican HorticulturalHorticultural SocietySociety JanuaryJanuary // FebruaryFebruary 20112011 New Plants for 2011 Unusual Trees with Garden Potential The AHS’s River Farm: A Center of Horticulture Fragrant Annuals Legacies assume many forms hether making estate plans, considering W year-end giving, honoring a loved one or planting a tree, the legacies of tomorrow are created today. Please remember the American Horticultural Society when making your estate and charitable giving plans. Together we can leave a legacy of a greener, healthier, more beautiful America. For more information on including the AHS in your estate planning and charitable giving, or to make a gift to honor or remember a loved one, please contact Courtney Capstack at (703) 768-5700 ext. 127. Making America a Nation of Gardeners, a Land of Gardens contents Volume 90, Number 1 . January / February 2011 FEATURES DEPARTMENTS 5 NOTES FROM RIVER FARM 6 MEMBERS’ FORUM 8 NEWS FROM THE AHS 2011 Seed Exchange catalog online for AHS members, new AHS Travel Study Program destinations, AHS forms partnership with Northeast garden symposium, registration open for 10th annual America in Bloom Contest, 2011 EPCOT International Flower & Garden Festival, Colonial Williamsburg Garden Symposium, TGOA-MGCA garden photography competition opens. 40 GARDEN SOLUTIONS Plant expert Scott Aker offers a holistic approach to solving common problems. 42 HOMEGROWN HARVEST page 28 Easy-to-grow parsley. 44 GARDENER’S NOTEBOOK Enlightened ways to NEW PLANTS FOR 2011 BY JANE BERGER 12 control powdery mildew, Edible, compact, upright, and colorful are the themes of this beating bugs with plant year’s new plant introductions. -

SCIENCE CHINA Complete Chloroplast Genome Sequence Of
SCIENCE CHINA Life Sciences • RESEARCH PAPER • February 2013 Vol.56 No.2: 189–198 doi: 10.1007/s11427-012-4430-8 Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species LI XiWen1,3†, GAO HuanHuan1,2†, WANG YiTao3, SONG JingYuan1, HENRY Robert4, WU HeZhen2, HU ZhiGang2, YAO Hui1, LUO HongMei1, LUO Kun1, PAN HongLin2 & CHEN ShiLin1,5* 1Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Beijing 100193, China; 2Faculty of Pharmacy, Hubei University of Chinese Medicine, Wuhan 430065, China; 3Institute of Chinese Medical Science, University of Macao, Macao 999078, China; 4Queensland Alliance for Agriculture and Food Innovation, University of Queensland, Brisbane QLD 4072, Australia; 5Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China Received July 13, 2012; accepted November 23, 2012; published online January 17, 2013 Magnolia grandiflora is an important medicinal, ornamental and horticultural plant species. The chloroplast (cp) genome of M. grandiflora was sequenced using a 454 sequencing platform and the genome structure was compared with other related species. The complete cp genome of M. grandiflora was 159623 bp in length and contained a pair of inverted repeats (IR) of 26563 bp separated by large and small single copy (LSC, SSC) regions of 87757 and 18740 bp, respectively. A total of 129 genes were successfully annotated, 18 of which included introns. The identity, number and GC content of M. grandiflora cp genes were similar to those of other Magnoliaceae species genomes. Analysis revealed 218 simple sequence repeat (SSR) loci, most com- posed of A or T, contributing to a bias in base composition. -

The Progressive and Ancestral Traits of the Secondary Xylem Within Magnolia Clad – the Early Diverging Lineage of Flowering Plants
Acta Societatis Botanicorum Poloniae ORIGINAL RESEARCH PAPER Acta Soc Bot Pol 84(1):87–96 DOI: 10.5586/asbp.2014.028 Received: 2014-07-31 Accepted: 2014-12-01 Published electronically: 2015-01-07 The progressive and ancestral traits of the secondary xylem within Magnolia clad – the early diverging lineage of flowering plants Magdalena Marta Wróblewska* Department of Developmental Plant Biology, Institute of Experimental Biology, University of Wrocław, Kanonia 6/8, 50-328 Wrocław, Poland Abstract The qualitative and quantitative studies, presented in this article, on wood anatomy of various species belonging to ancient Magnolia genus reveal new aspects of phylogenetic relationships between the species and show evolutionary trends, known to increase fitness of conductive tissues in angiosperms. They also provide new examples of phenotypic plasticity in plants. The type of perforation plate in vessel members is one of the most relevant features for taxonomic studies. InMagnolia , until now, two types of perforation plates have been reported: the conservative, scalariform and the specialized, simple one. In this paper, are presented some findings, new to magnolia wood science, like exclusively simple perforation plates in some species or mixed perforation plates – simple and scalariform in one vessel member. Intravascular pitting is another taxonomically important trait of vascular tissue. Interesting transient states between different patterns of pitting in one cell only have been found. This proves great flexibility of mechanisms, which elaborate cell wall structure in maturing trache- ary element. The comparison of this data with phylogenetic trees, based on the fossil records and plastid gene expression, clearly shows that there is a link between the type of perforation plate and the degree of evolutionary specialization within Magnolia genus. -

WO 2010/002406 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 7 January 2010 (07.01.2010) WO 2010/002406 Al (51) International Patent Classification: EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, A61K 36/00 (2006.01) HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, PCT/US2008/069017 NZ, OM, PG, PH, PL, PT, RO, RS, RU, SC, SD, SE, SG, (22) International Filing Date: SK, SL, SM, SV, SY, TJ, TM, TN, TR, TT, TZ, UA, UG, 2 July 2008 (02.07.2008) US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, (71) Applicant and ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, (72) Inventor: JAFFE, Russell, M. [US/US]; 10430 Hunter TM), European (AT, BE, BG, CH, CY, CZ, DE, DK, EE, View Road, Vienna, VA 22181 (US). ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, MC, MT, NL, NO, PL, PT, RO, SE, SI, SK, TR), OAPI (74) Agents: WARREN, Leigh, M. et al; Cooley Godward (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, ML, MR, Kronish LLP, 777 6th Street, N.W., Suite 1100, Washing NE, SN, TD, TG).