Actinin-4 Reveals a Mechanism for Regulating Its F-Actin-Binding Affinity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Spectrin Binding Motifs Control Scribble Cortical Dynamics And
1 2 3 Spectrin binding motifs control Scribble cortical 4 dynamics and polarity function 5 6 Batiste Boëda and Sandrine Etienne Manneville 7 8 Institut Pasteur (CNRS URA 3691-INSERM), France 9 Correspondance to Sandrine Etienne-Manneville. Cell Polarity, Migration and Cancer Unit, 10 Institut Pasteur, 25 rue du Dr Roux, 75724 Paris cedex 15, France; Phone: +33 1 4438 9591; 11 FAX: +33 1 4568 8548. 12 e-mail: [email protected] 13 14 Competing interests statement: 15 The authors declare that no competing interests exist. 16 17 1 18 Abstract 19 The tumor suppressor protein Scribble (SCRIB) plays an evolutionary conserved role in 20 cell polarity. Despite being central for its function, the molecular basis of SCRIB 21 recruitment and stabilization at the cell cortex is poorly understood. Here we show that 22 SCRIB binds directly to the CH1 domain of spectrins, a molecular scaffold that 23 contributes to the cortical actin cytoskeleton and connects it to the plasma membrane. 24 We have identified a short evolutionary conserved peptide motif named SADH motif 25 (SCRIB ABLIMs DMTN Homology) which is necessary and sufficient to mediate protein 26 interaction with spectrins. The SADH domains contribute to SCRIB dynamics at the 27 cell cortex and SCRIB polarity function. Furthermore, mutations in SCRIB SADH 28 domains associated with spina bifida and cancer impact the stability of SCRIB at the 29 plasma membrane, suggesting that SADH domain alterations may participate in human 30 pathology. 31 32 33 34 35 36 37 2 38 Introduction 39 The protein SCRIB has been implicated in a staggering array of cellular processes 40 including polarity, migration, proliferation, differentiation, apoptosis, stemcell 41 maintenance, and vesicle trafficking [1]. -

Current Understanding of the Role of Cytoskeletal Cross-Linkers in the Onset and Development of Cardiomyopathies
International Journal of Molecular Sciences Review Current Understanding of the Role of Cytoskeletal Cross-Linkers in the Onset and Development of Cardiomyopathies Ilaria Pecorari 1, Luisa Mestroni 2 and Orfeo Sbaizero 1,* 1 Department of Engineering and Architecture, University of Trieste, 34127 Trieste, Italy; [email protected] 2 University of Colorado Cardiovascular Institute, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA; [email protected] * Correspondence: [email protected]; Tel.: +39-040-5583770 Received: 15 July 2020; Accepted: 10 August 2020; Published: 15 August 2020 Abstract: Cardiomyopathies affect individuals worldwide, without regard to age, sex and ethnicity and are associated with significant morbidity and mortality. Inherited cardiomyopathies account for a relevant part of these conditions. Although progresses have been made over the years, early diagnosis and curative therapies are still challenging. Understanding the events occurring in normal and diseased cardiac cells is crucial, as they are important determinants of overall heart function. Besides chemical and molecular events, there are also structural and mechanical phenomena that require to be investigated. Cell structure and mechanics largely depend from the cytoskeleton, which is composed by filamentous proteins that can be cross-linked via accessory proteins. Alpha-actinin 2 (ACTN2), filamin C (FLNC) and dystrophin are three major actin cross-linkers that extensively contribute to the regulation of cell structure and mechanics. Hereby, we review the current understanding of the roles played by ACTN2, FLNC and dystrophin in the onset and progress of inherited cardiomyopathies. With our work, we aim to set the stage for new approaches to study the cardiomyopathies, which might reveal new therapeutic targets and broaden the panel of genes to be screened. -

CH Domains Revisited
View metadata,FEBS Letters citation 431 and (1998) similar 134^137 papers at core.ac.uk broughtFEBS to you 20514 by CORE provided by Elsevier - Publisher Connector Minireview CH domains revisited Theresia Stradala, Wolfgang Kranewittera, Steven J. Winderb, Mario Gimonaa;* aInstitute of Molecular Biology, Austrian Academy of Sciences, Department of Cell Biology, Billrothstrasse 11, A-5020 Salzburg, Austria bInstitute of Cell and Molecular Biology, University of Edinburgh, May¢eld Road, Edinburgh EH9 3JR, UK Received 2 June 1998 responding to one CH domain [9^11]. Together, this led to the Abstract A sequence motif of about 100 amino acids, termed the `calponin homology domain' has been suggested to confer commonly accepted misconception that CH domains function actin binding to a variety of cytoskeletal and signalling as an autonomous F-actin binding domain, and the CH do- molecules. Here we analyse and compare the sequences of all main was installed as the prototype for a novel actin-binding calponin homology domain-containing proteins identified to date. module [3,4]. Closer inspection of CH domain sequences, We propose that single calponin homology domains do not confer however, reveals that this region does not overlap the estab- actin-binding per se and that the actin-binding motifs of cross- lished actin-binding domain in calponin, delineated in several linking proteins, which comprise two disparate calponin homol- independent studies [12^14]. Moreover, a second actin-target- ogy domains, represent a unique protein module. ing site was recently identi¢ed in the C-terminal tandem re- z 1998 Federation of European Biochemical Societies. peats of calponin [15], and it was further demonstrated that the single CH domain is neither su¤cient nor necessary for F- Key words: Calponin; Calponin homology domain; Actin binding actin binding of calponin and SM22 [16]. -

Ankyrins, Spectrins, and Their Functional Patterning of Neurons
Cargo hold and delivery: Ankyrins, spectrins, and their functional patterning of neurons Damaris N. Lorenzo Department of Cell Biology and Physiology, The University of North Carolina at Chapel Abstract Hill, Chapel Hill, North Carolina The highly polarized, typically very long, and nonmitotic nature of neurons present Correspondence them with unique challenges in the maintenance of their homeostasis. This architec- Damaris N. Lorenzo, Department of Cell tural complexity serves a rich and tightly controlled set of functions that enables their Biology and Physiology, The University of North Carolina at Chapel Hill, Chapel Hill, NC fast communication with neighboring cells and endows them with exquisite plasticity. 27599. The submembrane neuronal cytoskeleton occupies a pivotal position in orchestrating Email: [email protected] the structural patterning that determines local and long-range subcellular specializa- Funding information tion, membrane dynamics, and a wide range of signaling events. At its center is the National Institute of Neurological Disorders and Stroke, Grant/Award Number: partnership between ankyrins and spectrins, which self-assemble with both remark- R01NS110810 able long-range regularity and micro- and nanoscale specificity to precisely position and stabilize cell adhesion molecules, membrane transporters, ion channels, and other cytoskeletal proteins. To accomplish these generally conserved, but often functionally divergent and spatially diverse, roles these partners use a combinatorial program of a couple of dozens interacting family members, whose code is not fully unraveled. In a departure from their scaffolding roles, ankyrins and spectrins also enable the delivery of material to the plasma membrane by facilitating intracellular transport. Thus, it is unsur- prising that deficits in ankyrins and spectrins underlie several neurodevelopmental, neu- rodegenerative, and psychiatric disorders. -

Actin-Bundling Proteins in Cancer Progression at a Glance
Cell Science at a Glance 1073 Actin-bundling proteins cellular structures, such as filopodia general, crosslinking proteins have two (spike-like protrusions), lamellipodia actin-binding sites, often because they in cancer progression at (sheet-like protrusions), stress fibers (elastic dimerise, and the location of actin-binding a glance contractile bundles), microvilli (finger-like sites determines the filament arrangement surface protrusions) and invadopodia and type of crosslinked structure formed. (invasive cell feet) (see Table 1 for a more Actin filaments are polar, with a fast- Richard P. Stevenson, Douwe complete list). Whereas the cytoskeleton is growing and a slow-growing end, and this Veltman and Laura M. Machesky* important in normal cellular function, it can polarity is maintained by a cycle of ATP The Beatson Institute for Cancer Research, Garscube be subverted in cancer cells and contributes hydrolysis (see Poster) (Pollard and Cooper, Estate, Switchback Rd, Bearsden, Glasgow G61 to changes in cell growth, stiffness, 2009). Bundling proteins can be selective 1BD, UK *Author for correspondence movement and invasiveness. We hereby about the orientation with which they bind to ([email protected]) give an overview of the role of actin- the filament, allowing the specific formation filament bundling in cellular structures and of bundles of either mixed or uniform Journal of Cell Science 125, 1073–1079 ß 2012. Published by The Company of Biologists Ltd discuss how alterations in the activity or polarity (see Poster). Bundling proteins are doi: 10.1242/jcs.093799 expression patterns of actin-bundling often modular and contain repeated actin- proteins could be linked to cancer initiation filament-binding domains (see Poster). -

Solution Structure of the Calponin CH Domain and Fitting to the 3D-Helical Reconstruction of F-Actin:Calponin
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Structure, Vol. 10, 249–258, February, 2002, 2002 Elsevier Science Ltd. All rights reserved. PII S0969-2126(02)00703-7 Solution Structure of the Calponin CH Domain and Fitting to the 3D-Helical Reconstruction of F-Actin:Calponin Janice Bramham,1,6 Julie L. Hodgkinson,2 tropomyosin, caldesmon, and calponin. Basic calponin, Brian O. Smith,3 Dusan Uhrı´n,4 Paul N. Barlow,3,4 one of three genetic variants found almost exclusively in and Steven J. Winder5 smooth muscle, was first isolated from chicken gizzard 1 Department of Biochemistry smooth muscle as a 34 kDa protein that binds to filamen- University of Leicester tous actin (F-actin) [1]. It has been proposed that cal- Adrian Building ponin has a role both in muscle contraction and as a University Road structural component in the cytoskeleton of smooth Leicester LE1 7RH muscle cells. Calponin is associated with the contractile United Kingdom apparatus, along with actin, myosin, and caldesmon. It 2 Imperial College School of Science also occurs in the membrane skeleton that forms the Technology and Medicine interface between the contractile apparatus and the ex- National Heart and Lung Institute tracellular matrix, and it appears in the cytoskeleton, Dovehouse Street which surrounds and supports the contractile appara- London SW3 6LY tus, along with -actin, filamin, and desmin [2]. United Kingdom In vitro studies have shown that calponin is an actin 3 Institute of Cell and Molecular Biology binding protein and a major regulator of muscle contrac- University of Edinburgh tion. -
Steric Regulation of Tandem Calponin Homology Domain Actin-Binding Affinity
bioRxiv preprint doi: https://doi.org/10.1101/598359; this version posted May 27, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. TITLE Steric Regulation of Tandem Calponin Homology Domain Actin-Binding Affinity AUTHORS Andrew R Harris1, Brian Belardi1, Pamela Jreij1, Kathy Wei1, Andreas Bausch3, Daniel A Fletcher1,2,4* AFFILIATIONS 1Department of Bioengineering and Biophysics Program, University of California, Berkeley, 648 Stanley Hall MC 1762, Berkeley, CA 94720, USA 2Biological Systems and Engineering Division, Lawrence Berkeley National Laboratory, 648 Stanley Hall MC 1762, Berkeley, CA 94720, USA 3Lehrstuhl für Biophysik (E27), Technische Universität München, Garching 85748, Germany. 4Chan Zuckerberg Biohub, San Francisco, CA 94158 *Daniel A Fletcher, [email protected] KEYWORDS Calponin Homology Domain / Actin / Cytoskeleton / Affinity RUNNING TITLE Steric CH1-CH2 regulation 1 bioRxiv preprint doi: https://doi.org/10.1101/598359; this version posted May 27, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT Tandem calponin homology domains are common actin-binding motifs found in proteins such as α-actinin, filamin, utrophin, and dystrophin that organize actin filaments into functional structures. Despite a conserved structural fold and regions of high sequence similarity, tandem calponin homology (CH1-CH2) domains can have remarkably different actin-binding properties, with disease-associated point mutants known to cause increased as well as decreased affinity for f-actin. How small variations in sequence can lead to significant differences in binding affinity and resulting actin organization remains unclear. -

2111396504.Pdf
Biochimica et Biophysica Acta 1838 (2014) 620–634 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbamem Review Spectrins: A structural platform for stabilization and activation of membrane channels, receptors and transporters☆ Beata Machnicka b,1, Aleksander Czogalla a,1, Anita Hryniewicz-Jankowska a,Dżamila M. Bogusławska b, Renata Grochowalska b,Elżbieta Heger b, Aleksander F. Sikorski a,⁎ a University of Wrocław, Biotechnology Faculty, Poland b University of Zielona Góra, Faculty of Biological Sciences, Poland article info abstract Article history: This review focuses on structure and functions of spectrin as a major component of the membrane skeleton. Received 31 January 2013 Recent advances on spectrin function as an interface for signal transduction mediation and a number of data Received in revised form 25 April 2013 concerning interaction of spectrin with membrane channels, adhesion molecules, receptors and transporters Accepted 6 May 2013 draw a picture of multifaceted protein. Here, we attempted to show the current depiction of multitask role of Available online 11 May 2013 spectrin in cell physiology. This article is part of a Special Issue entitled: Reciprocal influences between cell cytoskeleton and membrane channels, receptors and transporters. Guest Editor: Jean Claude Hervé. Keywords: Spectrin © 2013 Elsevier B.V. All rights reserved. Spectrin-based membrane skeleton Plasma membrane domain Contents 1. Introduction ............................................................. -

Structural Study of the WH2 Family and Filamin: Implications for Actin Cytoskeleton Regulation
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 182 Structural Study of the WH2 Family and Filamin: Implications for Actin Cytoskeleton Regulation ADELEKE H. AGUDA ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6206 UPPSALA ISBN 91-554-6679-6 2006 urn:nbn:se:uu:diva-7188 ! "# $% & ' ( ) &* % + ,! + + -, , ./ + 01 2, 3 # !,1 4! 4 1 &*1 " " + , ' & / / 5 + 4 6 7! 1 4 1 )&1 %8 1 1 5"( 9:%%8:**$9:*1 6 , , ! , ! , ! + , 6 1 2, 6 !, + : ! .4-0 , ; ! ! ; < 1 2, 4- ! < , '6 :4, " , ! & .' &0 , , ! . 0 ! 1 5 , 3 6 3 , + , + ! 3 , , =8 & , ' & + (:'4"- , 1 ' , 3 , , ! ' & + ! , =8 < ! : ,1 2, , + , + , + 1 ' , < ' & + + /: ! : ! 1 2, /: ! 3 , , + + , > ' & + , + , , ! 3 ! + 1 ' , ! + & , ?: /: , + ! , + ! 3 1 ' , + ; 8:* + , 6 ! :+ 1 - @: ! , ! , , 3 , , + , 1 4 2, (:'4"- / , ! ' & - ! , ! "# $ % & $ % ' ()*$ $ +,-(.*/ $ A 46 1 4! &* 5""( *%:*&* 5"( 9:%%8:**$9:* :$)) ., BB 161B C D :$))0 Figure 1. Model of Thymosin E4 bound to actin A bit beyond perception’s reach I sometimes believe I see That life is two locked boxes, Each containing the other’s key Piet Hein (1905-1996). It is not the -
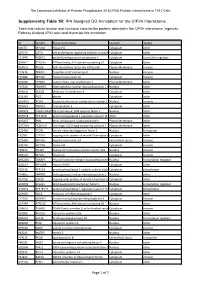
IPA Assigned GO Annotation for the CIP2A Interactome
The Cancerous Inhibitor of Protein Phosphatase 2A (CIP2A) Protein Interactome in Th17 Cells Supplementry Table 1B: IPA Assigned GO Annotation for the CIP2A Interactome Table lists cellular location and functional class for the proteins detected in the CIP2A interactome. Ingenuity Pathway Analysis (IPA) was used to provide this annotation ID Symbol Entrez Gene Name Location Type(s) B0I1T2 MYO1G myosin IG Cytoplasm other Q8TCG1 CIP2A cell proliferation regulating inhibitor of proteinCytoplasm phosphatase 2A other P11940 PABPC1 poly(A) binding protein cytoplasmic 1 Cytoplasm translation regulator Q9NVI7 ATAD3A ATPase family, AAA domain containing 3A Cytoplasm other Q9NVJ2 ARL8B ADP ribosylation factor like GTPase 8B Plasma Membrane enzyme P19474 TRIM21 tripartite motif containing 21 Nucleus enzyme P35580 MYH10 myosin heavy chain 10 Cytoplasm enzyme Q01082 SPTBN1 spectrin beta, non-erythrocytic 1 Plasma Membrane other P07910 HNRNPC heterogeneous nuclear ribonucleoprotein C (C1/C2)Nucleus other Q8NF50 DOCK8 dedicator of cytokinesis 8 Cytoplasm other Q15149 PLEC plectin Cytoplasm other Q9UKV3 ACIN1 apoptotic chromatin condensation inducer 1 Nucleus enzyme Q9NYL9 TMOD3 tropomodulin 3 Cytoplasm other P0DN76 U2AF1/U2AF1L5U2 small nuclear RNA auxiliary factor 1 Nucleus other Q6NYC8 PPP1R18 protein phosphatase 1 regulatory subunit 18 Other other Q9H307 PNN pinin, desmosome associated protein Plasma Membrane other Q7Z2W4 ZC3HAV1 zinc finger CCCH-type containing, antiviral 1 Plasma Membrane other Q13428 TCOF1 treacle ribosome biogenesis factor 1 Nucleus -

Structural Basis of the Filamin a Actin-Binding Domain Interaction with F-Actin
1 Structural basis of the filamin A actin-binding domain interaction with F-actin 2 Daniel V. Iwamoto1*, Andrew Huehn2*, Bertrand Simon1, Clotilde Huet-Calderwood1, 3 Massimiliano Baldassarre1,3, Charles V. Sindelar2† and David A. Calderwood1,4† 4 1Department of Pharmacology, 2Department of Molecular Biophysics and 5 Biochemistry, 4Department of Cell Biology, Yale University, New Haven Connecticut, 6 USA. 3University of Aberdeen Institute of Medical Sciences, Aberdeen, Scotland, UK. *These 7 authors contributed equally to this work. †Corresponding Authors, [email protected], 8 [email protected] 9 Abstract 10 Actin-crosslinking proteins assemble actin filaments into higher-order structures essential 11 for orchestrating cell shape, adhesion and motility. Missense mutations in the tandem 12 calponin homology (CH) domains of their actin-binding domains (ABDs) underlie numerous 13 genetic diseases, but a molecular understanding of these pathologies is hampered by the lack 14 of high-resolution structures of any actin-crosslinking protein bound to F-actin. Here, taking 15 advantage of a high-affinity, disease-associated mutant of the human filamin A (FLNa) ABD, 16 we combine cryo-electron microscopy and functional studies to reveal at near-atomic 17 resolution how the first CH domain (CH1) and residues immediately N-terminal to it engage 18 actin. We further show that reorientation of CH2 relative to CH1 is required to avoid clashes 19 with actin and to expose F-actin-binding residues on CH1. Our data explain localization of 20 disease-associated loss-of-function mutations to FLNaCH1 and gain-of-function mutations 21 to the regulatory FLNaCH2. Sequence conservation argues that this provides a general 22 model for ABD-F-actin binding. -

HISTOLOGY and HISTOPATHOLOGY (Non-Edited
ONLINE FIRST This is a provisional PDF only. Copyedited and fully formatted versión will Be made available at final puBliCation ISSN: 0213-3911 e-ISSN: 1699-5848 HISTOPATHOLOGY Submit your article to this Journal (http://www.hh.um.es/Instructions.htmmanuscript)) Membrane TraffickingAND and Exocytosis are Upregulated in Port Wine Stain Blood Vessels Authors: Rong Yin, Shawn J. Rice, Jinwei Wang, Lin Gao, Joseph Tsai, Radean T. Anvari, Fang Zhou, Xin Liu, Gang Wang, Yuxin Tang, Martin C. Mihm, Jr., Chandra P. Belani, Dong bao Chen, J. Stuart Nelson and Wenbin Tan (non-edited DOI: 10.14670/HH-18-051 Article type: ORIGINAL ARTICLE HISTOLOGYAccepted: 2018-10-10 Epub ahead of print: 2018-10-10 This artiCle has Been peer reviewed and puBlished immdediately upon acCeptanCe. ArtiCles in “Histology and Histopathology” are listed in PuBmed. Pre-print author´s version 1 Membrane Trafficking and Exocytosis are Upregulated in Port Wine Stain 2 Blood Vessels 3 Running Title: membrane trafficking and exocytosis in PWS 4 Rong Yin1,2,9, Shawn J. Rice3, Jinwei Wang1,8, Lin Gao5, Joseph Tsai1, Radean T. Anvari1, Fang 5 Zhou1,8, Xin Liu3, Gang Wang5, Yuxin Tang8, Martin C. Mihm, Jr.6, Chandra P. Belani3,4, Dong- 6 bao Chen10, J. Stuart Nelson1,7 and Wenbin Tan1,2* 7 1Department of Surgery, Beckman Laser Institute and Medical Clinic, University of California, 8 Irvine, Irvine, California 92617, USA 9 2Department of Cell Biology and Anatomy, University of South Carolina School of Medicine, 10 Columbia, South Carolina 29209, USA 11 3Penn State Cancer Institute, Hershey, PA 17033HISTOPATHOLOGY 12 4Department of Medicine, Penn State College of Medicine,manuscript) Hershey, PA 17033 13 5Department of Dermatology,AND Xijing Hospital, Xi'an, 710032, China.