Pi3kp110-, Src-, FAK-Dependent and DOCK2-Independent Migration and Invasion of CXCL13-Stimulated Prostate Cancer Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Structure of the Dock2âˆ'elmo1 Complex Provides Insights Into
ARTICLE https://doi.org/10.1038/s41467-020-17271-9 OPEN Structure of the DOCK2−ELMO1 complex provides insights into regulation of the auto-inhibited state Leifu Chang1,7, Jing Yang1, Chang Hwa Jo 2, Andreas Boland 1,8, Ziguo Zhang 1, Stephen H. McLaughlin 1, Afnan Abu-Thuraia 3, Ryan C. Killoran2, Matthew J. Smith 2,4,9, Jean-Francois Côté 3,5,6,9 & ✉ David Barford 1 DOCK (dedicator of cytokinesis) proteins are multidomain guanine nucleotide exchange 1234567890():,; factors (GEFs) for RHO GTPases that regulate intracellular actin dynamics. DOCK proteins share catalytic (DOCKDHR2) and membrane-associated (DOCKDHR1) domains. The structurally-related DOCK1 and DOCK2 GEFs are specific for RAC, and require ELMO (engulfment and cell motility) proteins for function. The N-terminal RAS-binding domain (RBD) of ELMO (ELMORBD) interacts with RHOG to modulate DOCK1/2 activity. Here, we determine the cryo-EM structures of DOCK2−ELMO1 alone, and as a ternary complex with RAC1, together with the crystal structure of a RHOG−ELMO2RBD complex. The binary DOCK2−ELMO1 complex adopts a closed, auto-inhibited conformation. Relief of auto- inhibition to an active, open state, due to a conformational change of the ELMO1 subunit, exposes binding sites for RAC1 on DOCK2DHR2, and RHOG and BAI GPCRs on ELMO1. Our structure explains how up-stream effectors, including DOCK2 and ELMO1 phosphorylation, destabilise the auto-inhibited state to promote an active GEF. 1 MRC Laboratory of Molecular Biology, Cambridge CB2 0QH, UK. 2 Institute for Research in Immunology and Cancer, Université de Montréal, Montréal, Québec H3T 1J4, Canada. 3 Montreal Institute of Clinical Research (IRCM), Montréal, QC H2W 1R7, Canada. -

CXCL13/CXCR5 Interaction Facilitates VCAM-1-Dependent Migration in Human Osteosarcoma
International Journal of Molecular Sciences Article CXCL13/CXCR5 Interaction Facilitates VCAM-1-Dependent Migration in Human Osteosarcoma 1, 2,3,4, 5 6 7 Ju-Fang Liu y, Chiang-Wen Lee y, Chih-Yang Lin , Chia-Chia Chao , Tsung-Ming Chang , Chien-Kuo Han 8, Yuan-Li Huang 8, Yi-Chin Fong 9,10,* and Chih-Hsin Tang 8,11,12,* 1 School of Oral Hygiene, College of Oral Medicine, Taipei Medical University, Taipei City 11031, Taiwan; [email protected] 2 Department of Orthopaedic Surgery, Chang Gung Memorial Hospital, Puzi City, Chiayi County 61363, Taiwan; [email protected] 3 Department of Nursing, Division of Basic Medical Sciences, and Chronic Diseases and Health Promotion Research Center, Chang Gung University of Science and Technology, Puzi City, Chiayi County 61363, Taiwan 4 Research Center for Industry of Human Ecology and Research Center for Chinese Herbal Medicine, Chang Gung University of Science and Technology, Guishan Dist., Taoyuan City 33303, Taiwan 5 School of Medicine, China Medical University, Taichung 40402, Taiwan; [email protected] 6 Department of Respiratory Therapy, Fu Jen Catholic University, New Taipei City 24205, Taiwan; [email protected] 7 School of Medicine, Institute of Physiology, National Yang-Ming University, Taipei City 11221, Taiwan; [email protected] 8 Department of Biotechnology, College of Health Science, Asia University, Taichung 40402, Taiwan; [email protected] (C.-K.H.); [email protected] (Y.-L.H.) 9 Department of Sports Medicine, College of Health Care, China Medical University, Taichung 40402, Taiwan 10 Department of Orthopedic Surgery, China Medical University Beigang Hospital, Yunlin 65152, Taiwan 11 Department of Pharmacology, School of Medicine, China Medical University, Taichung 40402, Taiwan 12 Chinese Medicine Research Center, China Medical University, Taichung 40402, Taiwan * Correspondence: [email protected] (Y.-C.F.); [email protected] (C.-H.T.); Tel.: +886-4-2205-2121-7726 (C.-H.T.); Fax: +886-4-2233-3641 (C.-H.T.) These authors contributed equally to this work. -

CXCL13/CXCR5 Signaling Axis in Cancer
Life Sciences 227 (2019) 175–186 Contents lists available at ScienceDirect Life Sciences journal homepage: www.elsevier.com/locate/lifescie Review article CXCL13/CXCR5 signaling axis in cancer T ⁎ Muzammal Hussaina,b,1, Dickson Adahb,c,1, Muqddas Tariqa,b, Yongzhi Lua, Jiancun Zhanga, , ⁎ Jinsong Liua, a Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, 190 Kaiyuan Avenue, Science Park, Guangzhou 510530, PR China b University of Chinese Academy of Sciences, Beijing 100049, PR China c State Key Laboratory of Respiratory Disease, Center for Infection and Immunity, Guangzhou Institutes of Biomedicine and Heath, Chinese Academy of Sciences, 190 Kaiyuan Avenue, Science Park, Guangzhou 510530, PR China ARTICLE INFO ABSTRACT Keywords: The tumor microenvironment comprises stromal and tumor cells which interact with each other through com- Cancer plex cross-talks that are mediated by a variety of growth factors, cytokines, and chemokines. The chemokine CXCL13 ligand 13 (CXCL13) and its chemokine receptor 5 (CXCR5) are among the key chemotactic factors which play CXCR5 crucial roles in deriving cancer cell biology. CXCL13/CXCR5 signaling axis makes pivotal contributions to the Tumor progression development and progression of several human cancers. In this review, we discuss how CXCL13/CXCR5 sig- Tumor immunity naling modulates cancer cell ability to grow, proliferate, invade, and metastasize. Furthermore, we also discuss Immune-evasion the preliminary evidence on context-dependent functioning of this axis within the tumor-immune micro- environment, thus, highlighting its potential dichotomy with respect to anticancer immunity and cancer im- mune-evasion mechanisms. At the end, we briefly shed light on the therapeutic potential or implications of targeting CXCL13/CXCR5 axis within the tumor microenvironment. -
HCC and Cancer Mutated Genes Summarized in the Literature Gene Symbol Gene Name References*
HCC and cancer mutated genes summarized in the literature Gene symbol Gene name References* A2M Alpha-2-macroglobulin (4) ABL1 c-abl oncogene 1, receptor tyrosine kinase (4,5,22) ACBD7 Acyl-Coenzyme A binding domain containing 7 (23) ACTL6A Actin-like 6A (4,5) ACTL6B Actin-like 6B (4) ACVR1B Activin A receptor, type IB (21,22) ACVR2A Activin A receptor, type IIA (4,21) ADAM10 ADAM metallopeptidase domain 10 (5) ADAMTS9 ADAM metallopeptidase with thrombospondin type 1 motif, 9 (4) ADCY2 Adenylate cyclase 2 (brain) (26) AJUBA Ajuba LIM protein (21) AKAP9 A kinase (PRKA) anchor protein (yotiao) 9 (4) Akt AKT serine/threonine kinase (28) AKT1 v-akt murine thymoma viral oncogene homolog 1 (5,21,22) AKT2 v-akt murine thymoma viral oncogene homolog 2 (4) ALB Albumin (4) ALK Anaplastic lymphoma receptor tyrosine kinase (22) AMPH Amphiphysin (24) ANK3 Ankyrin 3, node of Ranvier (ankyrin G) (4) ANKRD12 Ankyrin repeat domain 12 (4) ANO1 Anoctamin 1, calcium activated chloride channel (4) APC Adenomatous polyposis coli (4,5,21,22,25,28) APOB Apolipoprotein B [including Ag(x) antigen] (4) AR Androgen receptor (5,21-23) ARAP1 ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 1 (4) ARHGAP35 Rho GTPase activating protein 35 (21) ARID1A AT rich interactive domain 1A (SWI-like) (4,5,21,22,24,25,27,28) ARID1B AT rich interactive domain 1B (SWI1-like) (4,5,22) ARID2 AT rich interactive domain 2 (ARID, RFX-like) (4,5,22,24,25,27,28) ARID4A AT rich interactive domain 4A (RBP1-like) (28) ARID5B AT rich interactive domain 5B (MRF1-like) (21) ASPM Asp (abnormal -

Structural Basis for Mutual Relief of the Rac Guanine Nucleotide Exchange Factor DOCK2 and Its Partner ELMO1 from Their Autoinhibited Forms
Structural basis for mutual relief of the Rac guanine nucleotide exchange factor DOCK2 and its partner ELMO1 from their autoinhibited forms Kyoko Hanawa-Suetsugua, Mutsuko Kukimoto-Niinoa,b, Chiemi Mishima-Tsumagaria, Ryogo Akasakaa, Noboru Ohsawaa, Shun-ichi Sekinea,c, Takuhiro Itoa,c, Naoya Tochioa, Seizo Koshibaa, Takanori Kigawaa,d, Takaho Teradaa,b, Mikako Shirouzua, Akihiko Nishikimib,e,f, Takehito Urunoe, Tomoya Katakaig, Tatsuo Kinashig, Daisuke Kohdah, Yoshinori Fukuib,e,f,1, and Shigeyuki Yokoyamaa,b,c,1 aRIKEN Systems and Structural Biology Center, 1-7-22 Suehiro, Tsurumi, Yokohama 230-0045, Japan; bJapan Science and Technology Agency, Core Research for Evolutional Science and Technology, Tokyo 102-0075, Japan; cDepartment of Biophysics and Biochemistry, Graduate School of Science, University of Tokyo, 7-3-1 Hongo, Bunkyo, Tokyo 113-0033, Japan; dTokyo Institute of Technology, 4259 Nagatsuta, Midori, Yokohama 226-8502, Japan; eDivision of Immunogenetics, Department of Immunobiology and Neuroscience, Medical Institute of Bioregulation, Kyushu University, Fukuoka 812-8582, Japan; fResearch Center for Advanced Immunology, Kyushu University, Fukuoka 812-8582, Japan; gDepartment of Molecular Genetics, Institute of Biomedical Science, Kansai Medical University, Osaka 570-8506, Japan; and hDivision of Structural Biology, Medical Institute of Bioregulation, Kyushu University, Fukuoka 812-8582, Japan Edited by John Kuriyan, University of California, Berkeley, CA, and approved December 31, 2011 (received for review September 6, 2011) DOCK2, a hematopoietic cell-specific, atypical guanine nucleotide for the Rho-family GTPases. There are 11 mammalian members exchange factor, controls lymphocyte migration through ras-related (DOCK180, DOCK2-11) of the CDM family. The CDM proteins C3 botulinum toxin substrate (Rac) activation. -

Chemokine Ligand 13 Expression Is Abundant in the Tumor Microenvironment and Indicates Poor Prognosis of Kidney Clear Cell Carcinoma
BIOCELL Tech Science Press 2021 45(3): 589-597 Chemokine Ligand 13 Expression is Abundant in the Tumor Microenvironment and Indicates Poor Prognosis of Kidney Clear Cell Carcinoma MENGDAN WU1;MENGYAO SUN1;QINHUAI LAI1;YIN LU1;YUYIN FU1;YUJIA PENG1;WEIRONG LAI1;LISHI ZENG1; SHENGYAN ZHAO1;YUYAN LI1;ZHIXIONG ZHANG1;XIAOFENG CHEN1;FAN QIAO1;YIWEN ZHANG1,*;SHIJIE ZHOU1,2,*; LANTU GOU1;JINLIANG YANG1,2 1 Department of Biotherapy and Cancer Center/Collaborative Innovation Center for Biotherapy, West China Hospital, Sichuan University, Chengdu, 610041, China 2 Guangdong Zhongsheng Pharmaceutical Co., Ltd., Shantou, 515041, China Key words: CXCL13, Kidney clear cell carcinoma, Gamma/delta T cells, DNA methylation, Unfavorable survival Abstract: The chemokine ligand 13-chemokine receptor 5 (CXCL13-CXCR5) axis has been characterized as a critical tumor-promoting signaling pathway in the tumor microenvironment (TME) in multiple types of solid tumors. In this study, we analyzed the expression profile of CXCL13 in kidney clear cell carcinoma (KIRC) and its correlation with tumor-infiltrating immune cells (TIICs). A monoclonal antibody against CXCL13 with high affinity and purity was generated in our lab for western blot and immunohistochemistry (IHC). Bioinformatic analysis was performed based on bulk-seq data from the Cancer Genome Atlas (TCGA)-KIRC and single-cell RNA-seq data from scRNASeqDB and PanglaoDB. Results showed that high CXCL13 expression in TME was associated with shorter progression-free survival (PFS), disease-specific survival (DSS), and overall survival (OS). KIRC cell lines, as well as several other cancer cell lines, had negative CXCL13 expression. IHC staining from the Human Protein Atlas (HPA) and our tissue array indicated that CXCL13 might be mainly expressed by TIICs, but not KIRC tumor cells. -

Inflammation in Innate and Adaptive Immune Mechanisms October 28 – 30, 2012 Hilton Grand Wailea Resort | Maui, Hawai’I Organizers: Tom Hamilton and Xiaoxia Li
45th Annual Meeting of The Society for Leukocyte Biology InflammatIon In Innate and adaptIve Immune mechanIsms October 28 – 30, 2012 Hilton Grand Wailea Resort | Maui, Hawai’i Organizers: Tom Hamilton and Xiaoxia Li abstracts Journal of Leukocyte Biology, Supplement 2012 www.leukocytebiology.org 45th AnnuAl Meeting of the Society for leukocyte Biology inflAMMAtion in innAte And AdAptive iMMune MechAniSMS grAnd WAileA, MAui, hAWAi’i ~ octoBer 28-30, 2012 Thank you to the supporters of the 2012 SLB Meeting! Journal of Leukocyte Biology Supplement 2012 ABSTRACTS 1 2 Alcohol and Drugs of Abuse Interaction with HIV/AIDS: Chronic Alcohol Consumption Increases Mortality in Sepsis Systems Biology Approach in the SIV-Infected Macaque Benyam P. Yoseph, Zhe Liang, Elise Breed, Kevin McConnell, Patricia E. Molina David M. Guidot, Michael Koval, Craig M. Coopersmith Comprehensive Alcohol Research Center, Louisiana State Emory University School of Medicine University Health Sciences Center, NOLA Introduction: Excessive alcohol abuse is a problem of particular The two most commonly used and abused drugs are alcohol concern in the intensive care unit (ICU), as the rate of morbidity and the cannabinoids. Alcohol and drugs of abuse have been and mortality in all patients admitted to ICU is 2-4 times greater demonstrated to alter host response to human immunodeficiency than in non-alcoholics. Sepsis is the leading cause of death in ICU. (HIV) infection; by affecting progression of infection, tissue The purpose of this study was to examine the pathophysiology of injury, and time to death. Several factors can be involved in chronic alcohol abuse in sepsis. this, those pertaining to the host response, as well as those Methods: FVB/N mice were given liquid ethanol diet (20% w/v) or related to the ability of the virus to integrate itself into the host water for 12 weeks. -

Identification of Two Signaling Submodules Within the Crkii/ELMO
Cell Death and Differentiation (2007) 14, 963–972 & 2007 Nature Publishing Group All rights reserved 1350-9047/07 $30.00 www.nature.com/cdd Identification of two signaling submodules within the CrkII/ELMO/Dock180 pathway regulating engulfment of apoptotic cells A-C Tosello-Trampont1,4, JM Kinchen1,2,4, E Brugnera1,3, LB Haney1, MO Hengartner2 and KS Ravichandran*,1 Removal of apoptotic cells is a dynamic process coordinated by ligands on apoptotic cells, and receptors and other signaling proteins on the phagocyte. One of the fundamental challenges is to understand how different phagocyte proteins form specific and functional complexes to orchestrate the recognition/removal of apoptotic cells. One evolutionarily conserved pathway involves the proteins cell death abnormal (CED)-2/chicken tumor virus no. 10 (CT10) regulator of kinase (Crk)II, CED-5/180 kDa protein downstream of chicken tumor virus no. 10 (Crk) (Dock180), CED-12/engulfment and migration (ELMO) and MIG-2/RhoG, leading to activation of the small GTPase CED-10/Rac and cytoskeletal remodeling to promote corpse uptake. Although the role of ELMO : Dock180 in regulating Rac activation has been well defined, the function of CED-2/CrkII in this complex is less well understood. Here, using functional studies in cell lines, we observe that a direct interaction between CrkII and Dock180 is not required for efficient removal of apoptotic cells. Similarly, mutants of CED-5 lacking the CED-2 interaction motifs could rescue engulfment and migration defects in CED-5 deficient worms. Mutants of CrkII and Dock180 that could not biochemically interact could colocalize in membrane ruffles. -

P-Rex1 Directly Activates Rhog to Regulate GPCR-Driven Rac Signalling and Actin Polarity in Neutrophils
ß 2014. Published by The Company of Biologists Ltd | Journal of Cell Science (2014) 127, 2589–2600 doi:10.1242/jcs.153049 RESEARCH ARTICLE P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils George Damoulakis1,*, Laure Gambardella1, Kent L. Rossman2, Campbell D. Lawson1, Karen E. Anderson1, Yoshinori Fukui3, Heidi C. Welch1, Channing J. Der2, Len R. Stephens1,` and Phillip T. Hawkins1,`,§ ABSTRACT different Rho GTPase subfamilies and isoforms as key to their function in different cellular and subcellular contexts (Hung et al., G-protein-coupled receptors (GPCRs) regulate the organisation of 2013; Sanz-Moreno et al., 2008). In addition to cell migration the actin cytoskeleton by activating the Rac subfamily of small during normal development and immunity, deregulation of these GTPases. The guanine-nucleotide-exchange factor (GEF) P-Rex1 processes drives cancer invasion and metastasis, and there is is engaged downstream of GPCRs and phosphoinositide 3-kinase strong evidence that mutations in Rac1 and Rac GEFs are (PI3K) in many cell types, and promotes tumorigenic signalling and associated with disease initiation and/or progression (Berger metastasis in breast cancer and melanoma, respectively. Although et al., 2012; Hodis et al., 2012; Lindsay et al., 2011; Nishihara P-Rex1-dependent functions have been attributed to its GEF activity et al., 2002; Sanz-Moreno et al., 2008). towards Rac1, we show that P-Rex1 also acts as a GEF for the G-protein-coupled receptors (GPCRs) are acute regulators of Rac-related GTPase RhoG, both in vitro and in GPCR-stimulated cell movement, but the mechanisms by which they regulate GTP/ primary mouse neutrophils. -

NLRP3 Inflammasome Induces Chemotactic Immune Cell Migration
NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis Makoto Inouea, Kristi L. Williamsb,c, Michael D. Gunnb, and Mari L. Shinoharaa,d,1 Departments of aImmunology, bMedicine/Cardiology, and dMolecular Genetics and Microbiology, and cSchool of Nursing, Duke University Medical Center, Durham, NC 27710 Edited* by Harvey Cantor, Dana-Farber Cancer Institute, Boston, MA, and approved May 16, 2012 (received for review February 1, 2012) The NLRP3 inflammasome is a multiprotein complex consisting of apoptosis-associated speck-like protein containing a carboxy- three kinds of proteins, NLRP3, ASC, and pro-caspase-1, and plays terminal CARD (ASC), and pro-caspase-1, and found in innate a role in sensing pathogens and danger signals in the innate im- immune cells, such as macrophages and DCs. Active NLRP3 mune system. The NLRP3 inflammasome is thought to be involved inflammasome processes pro–IL-1β and pro–IL-18 to produce in the development of experimental autoimmune encephalomyeli- mature IL-1β and IL-18, respectively. We and another group − − tis (EAE), an animal model of multiple sclerosis (MS). However, the reported that mice lacking genes for Nlrp3 or Asc (Nlrp3 / and − − mechanism by which the NLRP3 inflammasome induces EAE is not Asc / mice) are resistant to the development of EAE (6, 7), clear. In this study, we found that the NLRP3 inflammasome suggesting the association of the NLRP3 inflammasome with played a critical role in inducing T-helper cell migration into the EAE development. In MS plaques and/or cells from MS patients, CNS. To gain migratory ability, CD4+ T cells need to be primed by the expression of caspase-1, IL-1β, and IL-18 is elevated (8–10), NLRP3 inflammasome-sufficient antigen-presenting cells to up- suggesting the involvement of the NLRP3 inflammasome in MS regulate chemotaxis-related proteins, such as osteopontin, CCR2, pathogenicity. -

HIV-1 Nef Binds the DOCK2–ELMO1 Complex to Activate Rac and Inhibit Lymphocyte Chemotaxis
PLoS BIOLOGY HIV-1 Nef Binds the DOCK2–ELMO1 Complex to Activate Rac and Inhibit Lymphocyte Chemotaxis Ajit Janardhan1,2, Tomek Swigut1¤1, Brian Hill1,2¤2, Michael P. Myers1, Jacek Skowronski1,2* 1 Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, United States of America, 2 Program in Genetics and Medical Scientist Training Program, Stony Brook University, Stony Brook, New York, United States of America The infectious cycle of primate lentiviruses is intimately linked to interactions between cells of the immune system. Nef, a potent virulence factor, alters cellular environments to increase lentiviral replication in the host, yet the mechanisms underlying these effects have remained elusive. Since Nef likely functions as an adaptor protein, we exploited a proteomic approach to directly identify molecules that Nef targets to subvert the signaling machinery in T cells. We purified to near homogeneity a major Nef-associated protein complex from T cells and identified by mass spectroscopy its subunits as DOCK2–ELMO1, a key activator of Rac in antigen- and chemokine-initiated signaling pathways, and Rac. We show that Nef activates Rac in T cell lines and in primary T cells following infection with HIV-1 in the absence of antigenic stimuli. Nef activates Rac by binding the DOCK2–ELMO1 complex, and this interaction is linked to the abilities of Nef to inhibit chemotaxis and promote T cell activation. Our data indicate that Nef targets a critical switch that regulates Rac GTPases downstream of chemokine- and antigen-initiated signaling pathways. This interaction enables Nef to influence multiple aspects of T cell function and thus provides an important mechanism by which Nef impacts pathogenesis by primate lentiviruses. -
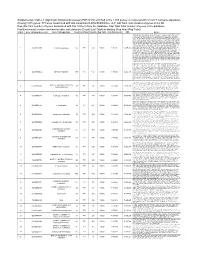
(FDR<0.05) Enriched in the 1015 Genes Co-Clustered with Known T
Supplementary Table 3. Significant biological processes (FDR<0.05) enriched in the 1,015 genes co-clustered with known T cell gene signatures. Among 1,015 genes, 771 were associated with GO annotation in DAVID database v6.7. List Total: total number of genes in my list. Pop Hits: total number of genes associated with this GO term from the database. Pop Total: total number of genes in the database. Fold Enrichment: relative enrichment ratio, calculated by (Count)/(List Total) divided by (Pop Hits)/(Pop Total). Index Gene Ontology Accession Gene Ontology Name Count List Total Pop Hits Pop Total Fold Enrichment FDR Genes AQP9, C1QC, B2M, LILRA1, LILRA2, CLEC4E, S1PR4, LILRA4, IFNG, LILRA6, CLEC4A, VNN1, ERAP2, FAS, CRTAM, C5AR1, GBP5, NCF2, NCF1, NCF4, SERPING1, HLA-DQA2, HLA-DQA1, PDCD1LG2, LILRB1, CCR9, C1QA, C1QB, LILRB2, CCR7, CCR6, UNC13D, CCR5, CD40LG, CCR4, LILRB3, CCR2, LILRB4, HLA-DPA1, VSIG4, HLA-DRA, IL1R2, IL1R1, HLA-DRB1, OAS3, ACP5, OAS1, OAS2, CD74, IFI35, ZAP70, FCER1G, HLA-DRB5, HLA-DPB1, HLA-DOA, HLA-DOB, DHX58, BLNK, IL23R, KIR2DS4, CD300C, SLAMF7, OASL, RGS1, APOL1, CD300A, HMHB1, CD209, CLEC7A, LY86, LY9, CLNK, FCRL4, SH2D1A, NOD2, HAMP, CCL3L1, CCL3L3, TICAM2, ICAM1, GZMA, CMKLR1, LY96, WAS, IL18BP, LAX1, TNFSF12- TNFSF13, HLA-DQB1, CSF2, GPR183, CCR1, GPR65, CXCL9, NCF1C, IL7R, CLEC10A, CCL24, CCL22, CYP27B1, CCL23, FCGR1C, FTHL3, FCGR1A, FCGR1B, BCL3, C2, CD27, CD28, FYB, IL18R1, IL7, CD1C, CTLA4, CCL19, CD1B, CD1A, TRIM22, CD180, CD1E, CCL18, CCL17, CCL13, FCGR2B, FCGR2C, P2RY14, LIME1, CD14, IL16, IL18, TLR1, TNFSF15,