Study Protocol Was Subject to Critical Review and Has Been Approved in the Present Version by the Undersigning Persons
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
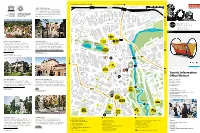
The Bauhaus and Weimar Modernism
Buchenwald Memorial, Ettersburg Castle Sömmerda (B7 / B85) 100 m weimar UNESCO World Heritage 500 m Culture City of Europe The Bauhaus and its sites in Weimar and Dessau have been on the UNESCO list of World Heritage since 1996. There are three objects in Weimar: the main building of the Bauhaus University Weimar, the former School of Applied Arts and the Haus Am Horn. Tiefurt Mansion deutschEnglish Harry-Graf-Kessler-Str. 10 5 Tiefurt Mansion Bauhaus-Universität Weimar Nietzsche Archive B Jorge-Semprùn-Platz a Oskar-Schlemmer-Str. d The building ensemble by Henry van de Velde was Friedrich Nietzsche spent the last years of his life at H e Stèphane- r 1 s revolutionary in terms of architecture at the turn of the “Villa Silberblick”. His sister established the Nietzsche Archive f Hessel-Platz e l d century. These Art School buildings became the venue here after his death and had the interior and furnishings e r S where the State Bauhaus was founded in 1919, making designed by Henry van de Velde. The current exhibition is t r a ß “Weimar” and the “Bauhaus” landmarks in the history of entitled “Kampf um Nietzsche” (“Dispute about Nietzsche”). e modern architecture. Humboldtstrasse 36 13 Mon, Wed to Sun 2pm – 5pm Geschwister-Scholl-Strasse 2 Mon to Fri 10am – 6pm | Sat & Sun 10am – 4pm Über dem Kegeltor C o u d r a y s t Erfurt (B7) r a ß e Berkaer Bahnhof 8 CRADLE, DESIGN: PETER KELER, 1922 © KLASSIK STIFTUNG WEIMAR 17 Jena (B7) 3 Tourist Information Office Weimar Haus Hohe Pappeln Weimar Municipal Museum 20 16 Markt 10, 99423 Weimar The Belgian architect Henry van de Velde, the artistic The permanent exhibition of the Municipal Museum presents Tel + 49 (0) 3643 745 0 advisor of the grand duchy, built this house for his family of “Democracy from Weimar. -

CHAPTER 2 the Period of the Weimar Republic Is Divided Into Three
CHAPTER 2 BERLIN DURING THE WEIMAR REPUBLIC The period of the Weimar Republic is divided into three periods, 1918 to 1923, 1924 to 1929, and 1930 to 1933, but we usually associate Weimar culture with the middle period when the post WWI revolutionary chaos had settled down and before the Nazis made their aggressive claim for power. This second period of the Weimar Republic after 1924 is considered Berlin’s most prosperous period, and is often referred to as the “Golden Twenties”. They were exciting and extremely vibrant years in the history of Berlin, as a sophisticated and innovative culture developed including architecture and design, literature, film, painting, music, criticism, philosophy, psychology, and fashion. For a short time Berlin seemed to be the center of European creativity where cinema was making huge technical and artistic strides. Like a firework display, Berlin was burning off all its energy in those five short years. A literary walk through Berlin during the Weimar period begins at the Kurfürstendamm, Berlin’s new part that came into its prime during the Weimar period. Large new movie theaters were built across from the Kaiser Wilhelm Memorial church, the Capitol und Ufa-Palast, and many new cafés made the Kurfürstendamm into Berlin’s avant-garde boulevard. Max Reinhardt’s theater became a major attraction along with bars, nightclubs, wine restaurants, Russian tearooms and dance halls, providing a hangout for Weimar’s young writers. But Berlin’s Kurfürstendamm is mostly famous for its revered literary cafés, Kranzler, Schwanecke and the most renowned, the Romanische Café in the impressive looking Romanische Haus across from the Memorial church. -

Energy and Environmental Technologies. Environmental Protection, Resource Efficiency, Green Tech – Key Technologies Made in Thuringia
09/2015 Energy and Environmental Technologies. Environmental protection, resource efficiency, green tech – key technologies made in Thuringia. Thuringian companies are among the world‘s leading providers of state-of-the-art power and environmental technologies: from conventional environmental protection and renewable energies to up-to-date technologies allowing an increase in energy efficiency. Quality made in Thuringia is in big demand, especially in waste Thuringia‘s energy and environmental technology processing, water and wastewater treatment, air pollution con- industry at a glance: trol, revitalization and renewable energies. By working closely > 366 companies with research institutions in these fields, Thuringia‘s companies > 5 research institutes can fully exploit their potential for growth. > 7 universities > leading engineering service providers in disciplines Proportion of companies such as industrial plant construction, hydrogeology, environmental geology and utilities (Source: In-house calculations according to LEG Industry/Technology Information Service, > market and technology leaders such as ENERCON, July 2013, N = 366 companies, multiple choices possible) Siemens and Vattenfall Seize the opportunities that our region offers. Benefit from a prime location in Europe’s heartland, highly skilled workers and a world-class research infrastructure. We provide full-service support for any investment project – from site search to project implementation and future expansions. Please contact us. www.invest-in-thuringia.de/en/top-industries/ environmental-technologies/ Skilled specialists – the keystone of success. Thuringia invests in the training and professional development of skilled workers so that your company can develop green, energy-efficient solutions for tomorrow. This maintains the competitiveness of Thuringian companies in these times of global climate change. -

Historical Aspects of Thuringia
Historical aspects of Thuringia Julia Reutelhuber Cover and layout: Diego Sebastián Crescentino Translation: Caroline Morgan Adams This publication does not represent the opinion of the Landeszentrale für politische Bildung. The author is responsible for its contents. Landeszentrale für politische Bildung Thüringen Regierungsstraße 73, 99084 Erfurt www.lzt-thueringen.de 2017 Julia Reutelhuber Historical aspects of Thuringia Content 1. The landgraviate of Thuringia 2. The Protestant Reformation 3. Absolutism and small states 4. Amid the restauration and the revolution 5. Thuringia in the Weimar Republic 6. Thuringia as a protection and defense district 7. Concentration camps, weaponry and forced labor 8. The division of Germany 9. The Peaceful Revolution of 1989 10. The reconstitution of Thuringia 11. Classic Weimar 12. The Bauhaus of Weimar (1919-1925) LZT Werra bridge, near Creuzburg. Built in 1223, it is the oldest natural stone bridge in Thuringia. 1. The landgraviate of Thuringia The Ludovingian dynasty reached its peak in 1040. The Wartburg Castle (built in 1067) was the symbol of the Ludovingian power. In 1131 Luis I. received the title of Landgrave (Earl). With this new political landgraviate groundwork, Thuringia became one of the most influential principalities. It was directly subordinated to the King and therefore had an analogous power to the traditional ducats of Bavaria, Saxony and Swabia. Moreover, the sons of the Landgraves were married to the aristocratic houses of the European elite (in 1221 the marriage between Luis I and Isabel of Hungary was consummated). Landgrave Hermann I. was a beloved patron of art. Under his government (1200-1217) the court of Thuringia was transformed into one of the most important centers for cultural life in Europe. -

Curriculum Vitae Germany
Historisches Institut Dr. Alexander Schmidt Akademischer Rat Universität Jena · Einrichtung · 07737 Jena Fürstengraben 13 07743 Jena Curriculum Vitae Germany Telefon: +49 36 41 9-4979 Telefax: +49 36 41 9-310 02 E-Mail: [email protected] ACADEMIC DEGREES Jena, 29. Februar 2020 M.A. (1,0, 2001, Friedrich Schiller University of Jena), Dr.phil. (summa cum laude, 2005, Friedrich Schiller University of Jena) ACADEMIC POSITION Akademischer Rat, Historisches Institut, Friedrich Schiller University of Jena PREVIOUS POSITIONS 07/2009-03/2017 Juniorprofessor of Intellectual History, Research Centre “Laboratory Enlightenment”, Friedrich Schiller University of Jena 10/2004-12/2008 Research fellow at the Collaborative Research Centre 482 “The Weimar-Jena phenomenon: Culture around 1800” (Sonderforschungsbereich 482 “Ereignis Weimar-Jena. Kultur um 1800”), Friedrich Schiller University of Jena 7/2001–1/2002 Research fellow at the Department of History (Early Modern Studies), Friedrich Schiller University of Jena GRANTS, AWARDS, SCHOLARSHIPS 7/2018 (together with Professor Paul Cheney, University of Chicago) Summer Institute for University and College Teachers sponsored by the National Endowment for the Humanities ($ 110,694), Visiting Faculty at University of Chicago 12/2017 Initial grant by the Centre for the Study of the Global Condition (Leipzig) for the “Human rights without religion?” project (5500 €) 7/2015-11/2016 Feodor-Lynen-Fellowship for Experienced Researchers (offered by the Alexander von Humboldt-Foundation) at the John U. -

Thuringia.Com
www.thats-thuringia.com That’s Thuringia. Ladies and Gentlemen, Thuringia is the region where successful collaboration between entrepreneurs and researchers goes back centuries. Looking to the future has been a long-standing tradition here. Just take Carl Zeiss, Ernst Abbe, and Otto Schott, who joined forces in Jena to lay the foundations for the modern optics industry and for a productive partnership between business and science. It’s a success story that the entrepreneurs and scientists in our Free State are continuing to write to this very day. And in the process, our producers and services providers can draw upon a multifaceted research environment which currently comprises no less than nine universities and universities of applied sciences, a total of 14 institutions run by the Fraunhofer, Leibniz, Max-Planck, and Helmholtz scientific societies, as well as eight research institutions with close ties to the economy. It’s the variety and the optimal mix of locational advantages that makes Thuringia so attractive for investors from all over the world. The central location of our Land at the heart of Germany will soon become even more of an advantage thanks to the new ICE high-speed train junction in Erfurt, which will significantly reduce travelling time to Berlin, Munich, and Frankfurt am Main. International companies seeking to locate to Thuringia can choose from our many top-notch industrial sites, which are situated along major highways and also include large-scale locations for those investors in need of more space. By now, Thuringia has surpassed Baden-Württem- berg as the Federal Land within Germany with the highest number of industrial operations per 100,000 inhabitants. -

Hans Haacke Biography
P A U L A C O O P E R G A L L E R Y Hans Haacke Biography 1936 Born Cologne, Germany 1956-60 Staatliche Werkakademie (State Art Academy), Kassel, Staatsexamen (equivalent of M.F.A.) 1960-61 Stanley William Hayter's Atelier 17, Paris 1961 Tyler School of Fine Arts, Temple University, Philadelphia 1962 Moves to New York 1963-65 Return to Cologne. Teaches at Pädagogische Hochschule, Kettwig, and other institutions 1966-67 Teaches at University of Washington, Seattle; Douglas College, Rutgers University, New Jersey; Philadelphia College of Art 1967 - 2002 Teaches at Cooper Union, New York (Professor of Art Emeritus) 1973 Guest Professorship, Hochschule für Bildende Künste, Hamburg 1979 Guest Professorship, Gesamthochschule, Essen 1994 Guest Professorship, Hochschule für Bildende Künste, Hamburg 1997 Regents Lecturer, University of California, Berkeley Lives in New York (since 1965) Awards 1960 Deutscher Akademischer Austauschdienst (DAAD) 1961 Fulbright Fellowship 1973 John Simon Guggenheim Foundation Fellowship 1978 National Endowment for the Arts 1991 College Art Association Distinguished Artist Award for Lifetime Achievement Deutscher Kritikerpreis for 1990 Honorary Doctorate in Fine Arts, Oberlin College 1993 Golden Lion (shared with Nam June Paik), Venice Biennale 1997 Kurt-Eisner-Foundation, Munich Honorary Doctorate Bauhaus-Universität Weimar 2001 Prize of Helmut-Kraft-Stiftung, Stuttgart 2002 College Art Association Distinguished Teaching of Art Award 2004 Peter-Weiss-Preis, Bochum 2008 Honorary Doctorate, San Francisco Art Institute -

Introduction to the Weimar Republic
Handout Introduction to the Weimar Republic Directions: Write a D next to information about the Weimar Republic that represents characteristics of a democracy and an X next to information that describes problems or challenges for a democracy. From Monarchy to Democracy D X After World War I, Germany’s political leaders sought to transform Germany from a monarchy to a democracy, called the Weimar Republic (1918–1933). The Weimar Constitution divided power into three branches of government. Elections were held for the president and the Reichstag (the legislature), while the judicial branch was appointed. The Weimar Constitution D X Adopted on August 11, 1919, the new Weimar Constitution spelled out the “basic rights and ob- ligations” of government officials and the citizens they served. Most of those rights and obliga- tions had not existed in Germany under the kaiser, including equality before the law, freedom of religion, and privacy. Despite the inclusion of these rights in the Weimar Constitution, individual freedom was not fully protected. Old laws that denied freedoms continued, including laws that discriminated against homosexual men and “Gypsies” (the name, considered derogatory today, used to describe two groups of people called the Sinti and Roma). The Reichstag D X Germans voted for a party, rather than a candidate, to fill the Reichstag (the German legisla- ture). The elections determined the percentage of seats each party received in the Reichstag, but the parties themselves selected the individuals who filled each allotted seat. For example, if a party received 36% of the vote, they would get 36% of the seats in the Reichstag. -

Education in the Weimar Republic
Handout Education in the Weimar Republic Directions: As you are reading, annotate the text by completing the following steps: 1. Circle words that are unfamiliar. 2. Put a question mark (?) in the margin in places where you feel confused. 3. Answer the questions that follow the text. In the Weimar Republic, Germany’s schools remained centers of tradition. Most teachers were conservative, both in their way of teaching and in their politics, and many were anti-socialist and antisemitic. A young man known as Klaus describes his schooling in the 1920s: We were taught history as a series of facts. We had to learn dates, names, places of battles. Periods during which Germany won wars were emphasized. Periods during which Germany lost wars were sloughed over. We heard very little about World War I, except that the Versailles peace treaty was a disgrace, which someday, in some vague way, would be rectified. In my school, one of the best in Berlin, there were three courses in Greek and Roman history, four in medieval history, and not one in government. If we tried to relate ideas we got from literature or history to current events, our teachers changed the subject. I really don’t believe that anyone was deliberately trying to evade politics. Those teachers really seemed to think that what went on in the Greek and Roman Empires was more important than what was happening on the streets of Berlin and Munich. They considered any attempt to bring up current political questions a distraction . because we hadn’t done our homework. -

Das Haus Am Horn in Weimar – Bauhausstätte Und Weltkulturerbe: Bau, Nutzung Und Denkmalpflege
112 IV. World Heritage Sites of the 20th Century – German Case Studies Michael Siebenbrodt Das Haus am Horn in Weimar – Bauhausstätte und Weltkulturerbe: Bau, Nutzung und Denkmalpflege sem einzigen realisierten Bauhaus-Gebäude in Weimar wurden zahlreiche funktionelle, material-technische, technologische und ökologische Innovationen praktisch verwirklicht und mit einem Team von Mitgliedern aller Bauhauswerkstätten umgesetzt. In der Hochzeit der Inflation trat der Berliner Bauunter- nehmer Adolf Sommerfeld als Geldgeber in Erscheinung, für den Gropius mit dem Bauhaus gerade eine Holzvilla fertig gestellt hatte. Die Weimarer Gewerkschaften setzten beim Haus Am Horn einen großen Bauarbeiterstreik aus, damit dieses wichtige Gebäude zum geplanten Ausstel- lungstermin fertig gestellt werden konnte. Ein Jahr zuvor hatte Gropius im Auftrag des Weimarer Gewerkschafts- kartells mit einer Reihe von Bauhaus-Studenten das Märzgefallenen-Denkmal in Weimar zu Ehren der Opfer des Kapp-Putsches von 1920 übergeben. Das Versuchshaus Am Horn konstituiert ein räumliches und ideengeschichtliches Dreieck von Haus-Prototypen im Weimarer Park an der Ilm. Den Archetypus eines deutschen Hauses stellt das schindelgedeckte Garten- haus aus dem 17. Jahrhundert dar, das der Großherzog seinem Freund Johann Wolfgang Goethe geschenkt hatte. Von den Nazis mit ihrem kulturpolitischen Protagonisten Schulze-Naumburg wurde dieses Haus mit steilem Dach als typisch deutsche Bauform der „kulturbolschewisti- schen Wüstenarchitektur“ der Moderne entgegengesetzt. Dagegen stellt das Römische Haus von Johann August Arens aus dem Jahr 1792 am gegenüberliegenden Ufer der Ilm die Moderne des ausgehenden 18. Jahrhunderts vor, das klassizistische Ideal einer aufgeklärten Ge- sellschaft. Wenn sich der Großherzog in diesem seinem Lieblingshaus aufhielt, signalisierte er Goethe abends seinen Gesprächsbedarf durch Lichtzeichen mit einer Kerze – moderne Kommunikation zur Goethezeit. -

A Genealogy of Genocide in Francoist Spain
Genocide Studies and Prevention: An International Journal Volume 8 Issue 1 Article 6 November 2013 A Genealogy of Genocide in Francoist Spain Antonio Miguez Macho Marie Curie Fellow, Contemporary and American History Department, University of Santiago de Compostela Follow this and additional works at: https://scholarcommons.usf.edu/gsp Recommended Citation Miguez Macho, Antonio (2013) "A Genealogy of Genocide in Francoist Spain," Genocide Studies and Prevention: An International Journal: Vol. 8: Iss. 1: Article 6. DOI: http://dx.doi.org/10.5038/1911-9933.8.1.4 Available at: https://scholarcommons.usf.edu/gsp/vol8/iss1/6 This Article is brought to you for free and open access by the Open Access Journals at Scholar Commons. It has been accepted for inclusion in Genocide Studies and Prevention: An International Journal by an authorized editor of Scholar Commons. For more information, please contact [email protected]. A Genealogy of Genocide in Francoist Spain1 Antonio Miguez Macho Marie Curie Fellow, Contemporary and American History Department University of Santiago de Compostela Summary: The extermination that was associated with the violence of the Spanish Civil War period and the early 1940s has been studied in depth in recent decades. Until now, however, the concept of genocide has not been discussed with an eye to understanding and interpreting this violence. The hermeneutical and comparative potential of the concept is, however, unquestionable. This article aims both to contextualize the origin and development of the debates about the concept of genocide, and to show what the concept could add to the debate in the case of Spain. -

Consuming Beauty in the Weimar Republic: a Discussion of Youth, Cosmetics, and Power in Vicki Baum's Play Pariser Platz 13 (1930)
Studies in 20th & 21st Century Literature Volume 43 Issue 2 Article 47 December 2019 Consuming Beauty in the Weimar Republic: A Discussion of Youth, Cosmetics, and Power in Vicki Baum's play Pariser Platz 13 (1930) Victoria Vygodskaia - Rust Southeast Missouri State University, [email protected] Follow this and additional works at: https://newprairiepress.org/sttcl Part of the Film and Media Studies Commons, French and Francophone Literature Commons, German Literature Commons, Latin American Literature Commons, Modern Literature Commons, and the Spanish Literature Commons This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 4.0 License. Recommended Citation Vygodskaia - Rust, Victoria (2019) "Consuming Beauty in the Weimar Republic: A Discussion of Youth, Cosmetics, and Power in Vicki Baum's play Pariser Platz 13 (1930)," Studies in 20th & 21st Century Literature: Vol. 43: Iss. 2, Article 47. https://doi.org/10.4148/2334-4415.2064 This Article is brought to you for free and open access by New Prairie Press. It has been accepted for inclusion in Studies in 20th & 21st Century Literature by an authorized administrator of New Prairie Press. For more information, please contact [email protected]. Consuming Beauty in the Weimar Republic: A Discussion of Youth, Cosmetics, and Power in Vicki Baum's play Pariser Platz 13 (1930) Abstract Published in 1930, Vicki Baum’s play Pariser Platz 13: Eine Komödie aus dem Schönheitssalon engaged the readership with an unorthodox and thoroughly modern heroine: the successful owner of international beauty salons Helen Bross. Helen personified the wishes and dreams of Baum’s readers: Helen’s autonomy, both personal and financial, allowed her ot be an active consumer of modernity and its pleasures: travel, interaction with celebrities, and luxurious lodging.