Resonance Structures, Electron Mobility and Delocalization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aromaticity Sem- Ii
AROMATICITY SEM- II In 1931, German chemist and physicist Sir Erich Hückel proposed a theory to help determine if a planar ring molecule would have aromatic properties .This is a very popular and useful rule to identify aromaticity in monocyclic conjugated compound. According to which a planar monocyclic conjugated system having ( 4n +2) delocalised (where, n = 0, 1, 2, .....) electrons are known as aromatic compound . For example: Benzene, Naphthalene, Furan, Pyrrole etc. Criteria for Aromaticity 1) The molecule is cyclic (a ring of atoms) 2) The molecule is planar (all atoms in the molecule lie in the same plane) 3) The molecule is fully conjugated (p orbitals at every atom in the ring) 4) The molecule has 4n+2 π electrons (n=0 or any positive integer Why 4n+2π Electrons? According to Hückel's Molecular Orbital Theory, a compound is particularly stable if all of its bonding molecular orbitals are filled with paired electrons. - This is true of aromatic compounds, meaning they are quite stable. - With aromatic compounds, 2 electrons fill the lowest energy molecular orbital, and 4 electrons fill each subsequent energy level (the number of subsequent energy levels is denoted by n), leaving all bonding orbitals filled and no anti-bonding orbitals occupied. This gives a total of 4n+2π electrons. - As for example: Benzene has 6π electrons. Its first 2π electrons fill the lowest energy orbital, and it has 4π electrons remaining. These 4 fill in the orbitals of the succeeding energy level. The criteria for Antiaromaticity are as follows: 1) The molecule must be cyclic and completely conjugated 2) The molecule must be planar. -

Fundamental Studies of Early Transition Metal-Ligand Multiple Bonds: Structure, Electronics, and Catalysis
Fundamental Studies of Early Transition Metal-Ligand Multiple Bonds: Structure, Electronics, and Catalysis Thesis by Ian Albert Tonks In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy CALIFORNIA INSTITUTE OF TECHNOLOGY Pasadena, California 2012 Defended December 6th 2011 ii 2012 Ian A Tonks All Rights Reserved iii ACKNOWLEDGEMENTS I am extremely fortunate to have been surrounded by enthusiastic, dedicated, and caring mentors, colleagues, and friends throughout my academic career. A Ph.D. thesis is by no means a singular achievement; I wish to extend my wholehearted thanks to everyone who has made this journey possible. First and foremost, I must thank my Ph.D. advisor, Prof. John Bercaw. I think more so than anything else, I respect John for his character, sense of fairness, and integrity. I also benefitted greatly from John’s laissez-faire approach to guiding our research group; I’ve always learned best when left alone to screw things up, although John also has an uncanny ability for sensing when I need direction or for something to work properly on the high-vac line. John also introduced me to hiking and climbing in the Eastern Sierras and Owens Valley, which remain amongst my favorite places on Earth. Thanks for always being willing to go to the Pizza Factory in Lone Pine before and after all the group hikes! While I never worked on any of the BP projects that were spearheaded by our co-PI Dr. Jay Labinger, I must also thank Jay for coming to all of my group meetings, teaching me an incredible amount while I was TAing Ch154, and for always being willing to talk chemistry and answer tough questions. -

The Chemistry of Alkynes
14_BRCLoudon_pgs4-2.qxd 11/26/08 9:04 AM Page 644 14 14 The Chemistry of Alkynes An alkyne is a hydrocarbon containing a carbon–carbon triple bond; the simplest member of this family is acetylene, H C'C H. The chemistry of the carbon–carbon triple bond is similar in many respects toL that ofL the carbon–carbon double bond; indeed, alkynes and alkenes undergo many of the same addition reactions. Alkynes also have some unique chem- istry, most of it associated with the bond between hydrogen and the triply bonded carbon, the 'C H bond. L 14.1 NOMENCLATURE OF ALKYNES In common nomenclature, simple alkynes are named as derivatives of the parent compound acetylene: H3CCC' H L L methylacetylene H3CCC' CH3 dimethylacetyleneL L CH3CH2 CC' CH3 ethylmethylacetyleneL L Certain compounds are named as derivatives of the propargyl group, HC'C CH2 , in the common system. The propargyl group is the triple-bond analog of the allyl group.L L HC' C CH2 Cl H2CA CH CH2 Cl L L LL propargyl chloride allyl chloride 644 14_BRCLoudon_pgs4-2.qxd 11/26/08 9:04 AM Page 645 14.1 NOMENCLATURE OF ALKYNES 645 We might expect the substitutive nomenclature of alkynes to be much like that of alkenes, and it is. The suffix ane in the name of the corresponding alkane is replaced by the suffix yne, and the triple bond is given the lowest possible number. H3CCC' H CH3CH2CH2CH2 CC' CH3 H3C CH2 C ' CH L L L L L L L propyne 2-heptyne 1-butyne H3C CH C ' C CH3 HC' C CH2 CH2 C' C CH3 L L L L 1,5-heptadiyneLL L "CH3 4-methyl-2-pentyne Substituent groups that contain a triple bond (called alkynyl groups) are named by replac- ing the final e in the name of the corresponding alkyne with the suffix yl. -

Basic Concepts of Chemical Bonding
Basic Concepts of Chemical Bonding Cover 8.1 to 8.7 EXCEPT 1. Omit Energetics of Ionic Bond Formation Omit Born-Haber Cycle 2. Omit Dipole Moments ELEMENTS & COMPOUNDS • Why do elements react to form compounds ? • What are the forces that hold atoms together in molecules ? and ions in ionic compounds ? Electron configuration predict reactivity Element Electron configurations Mg (12e) 1S 2 2S 2 2P 6 3S 2 Reactive Mg 2+ (10e) [Ne] Stable Cl(17e) 1S 2 2S 2 2P 6 3S 2 3P 5 Reactive Cl - (18e) [Ar] Stable CHEMICAL BONDSBONDS attractive force holding atoms together Single Bond : involves an electron pair e.g. H 2 Double Bond : involves two electron pairs e.g. O 2 Triple Bond : involves three electron pairs e.g. N 2 TYPES OF CHEMICAL BONDSBONDS Ionic Polar Covalent Two Extremes Covalent The Two Extremes IONIC BOND results from the transfer of electrons from a metal to a nonmetal. COVALENT BOND results from the sharing of electrons between the atoms. Usually found between nonmetals. The POLAR COVALENT bond is In-between • the IONIC BOND [ transfer of electrons ] and • the COVALENT BOND [ shared electrons] The pair of electrons in a polar covalent bond are not shared equally . DISCRIPTION OF ELECTRONS 1. How Many Electrons ? 2. Electron Configuration 3. Orbital Diagram 4. Quantum Numbers 5. LEWISLEWIS SYMBOLSSYMBOLS LEWISLEWIS SYMBOLSSYMBOLS 1. Electrons are represented as DOTS 2. Only VALENCE electrons are used Atomic Hydrogen is H • Atomic Lithium is Li • Atomic Sodium is Na • All of Group 1 has only one dot The Octet Rule Atoms gain, lose, or share electrons until they are surrounded by 8 valence electrons (s2 p6 ) All noble gases [EXCEPT HE] have s2 p6 configuration. -

Early- Versus Late-Transition-Metal-Oxo Bonds: the Electronlc Structure of VO' and Ruo'
J. Phys. Chem. 1988, 92, 2109-2115 2109 Early- versus Late-Transition-Metal-Oxo Bonds: The Electronlc Structure of VO' and RuO' Emily A. Cartert and William A. Goddard III* Arthur Amos Noyes Laboratory of Chemical Physics,$ California Institute of Technology, Pasadena, California 91125 (Received: July 9, 1987; In Final Form: November 3, 1987) From all-electron ab initio generalized valence bond calculations (GVBCI-SCF) on VO+ and RuO', we find that an accurate description of the bonding is obtained only when important resonance configurations are included self-consistently in the wave function. The ground state of VO+('Z-) has a triple bond similar to that of CO, with D,""(V-O) = 128.3 kcal/mol [DFptl(V-O) = 1.31 * 5 kcal/mol], while the ground state of RuO+(~A)has a double bond similar to that of Oz, with D,CS'cd(Ru-O) = 67.1 kcal/mol. Vertical excitation energies for a number of low-lying electronic states of VO+ and RuO' are also reported. These results indicate fundamental differences in the nature of the metal-oxo bond in early and late metal oxo complexes that explain the observed trends in reactivity (e.g., early metal oxides are thermodynamically stable whereas late metal oxo complexes are highly reactive oxidants). Finally, we have used these results to predict the ground states of MO' for other first-row transition-metal oxides. I. Introduction TABLE I: First-Row Transition-Metal-Oxo Bond Strengths While the electronic structure of neutral transition-metal oxides (kcal/mol)' has been examined by several authors,' the only cationic tran- metal Do(M+-O) Do(M-0) metal Do(M+-O) Do(M-0) sition-metal oxide (TMO) which has been studied with correlated 3 Mn 3 wave functions is2 CrO+. -

Polar Covalent Bonds: Electronegativity
Polar Covalent Bonds: Electronegativity Covalent bonds can have ionic character These are polar covalent bonds Bonding electrons attracted more strongly by one atom than by the other Electron distribution between atoms is not symmetrical Bond Polarity and Electronegativity Symmetrical Covalent Bonds Polar Covalent Bonds C – C + - C – H C – O (non-polar) (polar) Electronegativity (EN): intrinsic ability of an atom to attract the shared electrons in a covalent bond Inductive Effect: shifting of sigma bonded electrons in resppygonse to nearby electronegative atom The Periodic Table and Electronegativity C – H C - Br and C - I (non-pol)lar) (po l)lar) Bond Polarity and Inductive Effect Nonpolar Covalent Bonds: atoms with similar EN Polar Covalent Bonds: Difference in EN of atoms < 2 Ionic Bonds: Difference in EN > 2 C–H bonds, relatively nonpolar C-O, C-X bonds (more electronegative elements) are polar Bonding electrons shift toward electronegative atom C acqqppuires partial positive char g,ge, + Electronegative atom acquires partial negative charge, - Inductive effect: shifting of electrons in a bond in response to EN of nearby atoms Electrostatic Potential Maps Electrostatic potential maps show calculated charge distributions Colors indicate electron- rich (red) and electron- poor (blue ) reg ions Arrows indicate direction of bond polarity Polar Covalent Bonds: Net Dipole Moments Molecules as a whole are often polar from vector summation of individual bond polarities and lone-pair contributions Strongly polar substances soluble in polar solvents like water; nonpolar substances are insoluble in water. Dipole moment ( ) - Net molecular polarity, due to difference in summed charges - magnitude of charge Q at end of molecular dipole times distance r between charges = Q r, in debyes (D), 1 D = 3.336 1030 coulomb meter length of an average covalent bond, the dipole moment would be 1.60 1029 Cm, or 4.80 D. -

Heterocycles 2 Daniel Palleros
Heterocycles 2 Daniel Palleros Heterocycles 1. Structures 2. Aromaticity and Basicity 2.1 Pyrrole 2.2 Imidazole 2.3 Pyridine 2.4 Pyrimidine 2.5 Purine 3. Π-excessive and Π-deficient Heterocycles 4. Electrophilic Aromatic Substitution 5. Oxidation-Reduction 6. DNA and RNA Bases 7. Tautomers 8. H-bond Formation 9. Absorption of UV Radiation 10. Reactions and Mutations Heterocycles 3 Daniel Palleros Heterocycles Heterocycles are cyclic compounds in which one or more atoms of the ring are heteroatoms: O, N, S, P, etc. They are present in many biologically important molecules such as amino acids, nucleic acids and hormones. They are also indispensable components of pharmaceuticals and therapeutic drugs. Caffeine, sildenafil (the active ingredient in Viagra), acyclovir (an antiviral agent), clopidogrel (an antiplatelet agent) and nicotine, they all have heterocyclic systems. O CH3 N HN O O N O CH 3 N H3C N N HN N OH O S O H N N N 2 N O N N O CH3 N CH3 caffeine sildenafil acyclovir Cl S N CH3 N N H COOCH3 nicotine (S)-clopidogrel Here we will discuss the chemistry of this important group of compounds beginning with the simplest rings and continuing to more complex systems such as those present in nucleic acids. Heterocycles 4 Daniel Palleros 1. Structures Some of the most important heterocycles are shown below. Note that they have five or six-membered rings such as pyrrole and pyridine or polycyclic ring systems such as quinoline and purine. Imidazole, pyrimidine and purine play a very important role in the chemistry of nucleic acids and are highlighted. -

AROMATIC COMPOUNDS Aromaticity
AROMATICITY AROMATIC COMPOUNDS Aromaticity Benzene - C6H6 H H H H H H H H H H H H Kekulé and the Structure of Benzene Kekule benzene: two forms are in rapid equilibrium 154 pm 134 pm • All bonds are 140 pm (intermediate between C-C and C=C) • C–C–C bond angles are 120° • Structure is planar, hexagonal A Resonance Picture of Bonding in Benzene resonance hybrid 6 -electron delocalized over 6 carbon atoms The Stability of Benzene Aromaticity: cyclic conjugated organic compounds such as benzene, exhibit special stability due to resonance delocalization of -electrons. Heats of hydrogenation + H2 + 120 KJ/mol + 2 H2 + 230 KJ/mol calc'd value= 240 KJ/mol 10 KJ/mol added stability + 3 H 2 + 208 KJ/mol calc'd value= 360 KJ/mol 152 KJ/mol added stability + 3 H2 + 337 KJ/mol 1,3,5-Hexatriene - conjugated but not cyclic Resonance energy of benzene is 129 - 152 KJ/mol An Orbital Hybridization View of Bonding in Benzene • Benzene is a planar, hexagonal cyclic hydrocarbon • The C–C–C bond angles are 120° = sp2 hybridized • Each carbon possesses an unhybridized p-orbital, which makes up the conjugated -system. • The six -electrons are delocalized through the -system The Molecular Orbitals of Benzene - the aromatic system of benzene consists of six p-orbitals (atomic orbitals). Benzene must have six molecular orbitals. Y6 Y4 Y5 six p-orbitals Y2 Y3 Y1 Degenerate orbitals: Y : zero nodes orbitals that have the 1 Bonding same energy Y2 and Y3: one node Y and Y : two nodes 4 5 Anti-bonding Y6: three node Substituted Derivatives of Benzene and Their Nomenclature -

Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc
Metal-Metal (MM) Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc Richard H. Duncan Lyngdoh*,a, Henry F. Schaefer III*,b and R. Bruce King*,b a Department of Chemistry, North-Eastern Hill University, Shillong 793022, India B Centre for Computational Quantum Chemistry, University of Georgia, Athens GA 30602 ABSTRACT: This survey of metal-metal (MM) bond distances in binuclear complexes of the first row 3d-block elements reviews experimental and computational research on a wide range of such systems. The metals surveyed are titanium, vanadium, chromium, manganese, iron, cobalt, nickel, copper, and zinc, representing the only comprehensive presentation of such results to date. Factors impacting MM bond lengths that are discussed here include (a) n+ the formal MM bond order, (b) size of the metal ion present in the bimetallic core (M2) , (c) the metal oxidation state, (d) effects of ligand basicity, coordination mode and number, and (e) steric effects of bulky ligands. Correlations between experimental and computational findings are examined wherever possible, often yielding good agreement for MM bond lengths. The formal bond order provides a key basis for assessing experimental and computationally derived MM bond lengths. The effects of change in the metal upon MM bond length ranges in binuclear complexes suggest trends for single, double, triple, and quadruple MM bonds which are related to the available information on metal atomic radii. It emerges that while specific factors for a limited range of complexes are found to have their expected impact in many cases, the assessment of the net effect of these factors is challenging. -
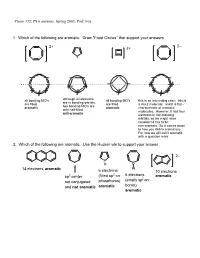
PS 6 Answers, Spring 2003, Prof
Chem 332, PS 6 answers, Spring 2003, Prof. Fox 1. Which of the following are aromatic. Draw "Frost Circles" that support your answers 2– 2+ 2+ + all bonding MO's although all electrons all bonding MO's this is an interesting case: this is are in bonding orbitals, are filled are filled a 4n+2 molecule, and it is flat-- two bonding MO's are aromatic aromatic characteristic of aromatic only half-filled molecules. However, it has four anti-aromatic electrons in non-bonding orbitals, so we might have considered this to be non-aromatic. So it comes down to how you define aromaticity. For now we will call it aromatic with a question mark. 2. Which of the following are aromatic. Use the Huckel rule to support your answer 2– P H B 14 electrons, aromatic H 6 electrons 10 electrons 2 6 electrons sp3 center (filled sp on aromatic (empty sp2 on not conjugated phosphorus) boron) and not aromatic aromatic aromatic 3. Compound 1 has an unusually large dipole moment for an organic hydrocarbon. Explain why (consider resonance structures for the central double bond) We need to remember that for any double bond compound, we can draw two polar resonance structure. Normally, these resonance structures are unimportant unimportant unimportant However, for 1 the story is different because there is a polar resonance form that has two aromatic components. In other words, 'breaking' the double bond gives rise to aromaticity, and therefore the polar resonance structure is important and leads to the observed dipole. dipole 1 8 e– 4 e– 6 e– 6 e– anti- anti- aromatic aromatic aromatic aromatic important resonance unimportant resonance structure structure 5. -

Variation of Aromaticity by Twisting Or Expanding the Ring Content*
Pure Appl. Chem., Vol. 82, No. 4, pp. 769–800, 2010. doi:10.1351/PAC-CON-09-11-07 © 2010 IUPAC, Publication date (Web): 26 March 2010 Variation of aromaticity by twisting or expanding the ring content* Remi Chauvin‡, Christine Lepetit, Valérie Maraval, and Léo Leroyer Laboratory of Coordination Chemistry (LCC), CNRS, 205, Route de Narbonne, F-31077 Toulouse, France; Université de Toulouse; UPS, INPT; LCC; F-31077 Toulouse, France Abstract: Generalization of the Hückel rule predicts that the (anti)aromaticity of a neutral ring is qualitatively reverted upon a single twist of the π-orbital array (Möbius interconver- sion), and is preserved upon expansion of all the bonds by single C2 units (ring carbo-mer- ization). These opposite effects are addressed from quantitative theoretical and experimental standpoints, respectively. (i) According to most resonance energy (RE) schemes, the RE value of a Möbius ring is not the opposite of that of the Hückel version. This also applies to the Aihara’s and Trinajstic’s topological resonance energy (TRE), where a non-aromatic ref- erence in the topological limit is defined as being “as identical as possible” to the parent ring but just “acyclic”. In spite of its conceptual merits, the computing complexity and fictitious character of the TRE acyclic reference resulted in a disuse of TRE as a current energetic aro- maticity index. Both the calculation and interpretation of TRE have been revisited in light of a cross-reference between the Hückel and Möbius rings within the Hückel molecular orbital (HMO) framework. Whereas the topological influence of triple bonds is currently neglected in the first-level HMO treatment of π-conjugated systems, a graph-theoretical analysis allows one to differentiate the TRE value of a [3n]annulene from those of the corresponding carbo- [n]annulene. -

Structure of Benzene, Orbital Picture, Resonance in Benzene, Aromatic Characters, Huckel’S Rule B
Dr.Mrityunjay Banerjee, IPT, Salipur B PHARM 3rd SEMESTER BP301T PHARMACEUTICAL ORGANIC CHEMISTRY –II UNIT I UNIT I Benzene and its derivatives 10 Hours A. Analytical, synthetic and other evidences in the derivation of structure of benzene, Orbital picture, resonance in benzene, aromatic characters, Huckel’s rule B. Reactions of benzene - nitration, sulphonation, halogenationreactivity, Friedelcrafts alkylation- reactivity, limitations, Friedelcrafts acylation. C. Substituents, effect of substituents on reactivity and orientation of mono substituted benzene compounds towards electrophilic substitution reaction D. Structure and uses of DDT, Saccharin, BHC and Chloramine Benzene and its Derivatives Chemists have found it useful to divide all organic compounds into two broad classes: aliphatic compounds and aromatic compounds. The original meanings of the words "aliphatic" (fatty) and "aromatic" (fragrant/ pleasant smell). Aromatic compounds are benzene and compounds that resemble benzene in chemical behavior. Aromatic properties are those properties of benzene that distinguish it from aliphatic hydrocarbons. Benzene: A liquid that smells like gasoline Boils at 80°C & Freezes at 5.5°C It was formerly used to decaffeinate coffee and component of many consumer products, such as paint strippers, rubber cements, and home dry-cleaning spot removers. A precursor in the production of plastics (such as Styrofoam and nylon), drugs, detergents, synthetic rubber, pesticides, and dyes. It is used as a solvent in cleaning and maintaining printing equipment and for adhesives such as those used to attach soles to shoes. Benzene is a natural constituent of petroleum products, but because it is a known carcinogen, its use as an additive in gasoline is now limited. In 1970s it was associated with leukemia deaths.