1 Optical Absorption of Excimer Laser-Induced Dichlorine Monoxide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
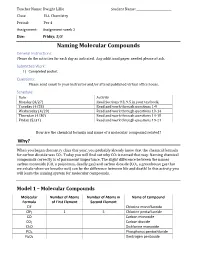
Naming Molecular Compounds General Instructions: Please Do the Activities for Each Day As Indicated
Teacher Name: Dwight Lillie Student Name: ________________________ Class: ELL Chemistry Period: Per 4 Assignment: Assignment week 2 Due: Friday, 5/8 Naming Molecular Compounds General Instructions: Please do the activities for each day as indicated. Any additional paper needed please attach. Submitted Work: 1) Completed packet. Questions: Please send email to your instructor and/or attend published virtual office hours. Schedule: Date Activity Monday (4/27) Read Sections 9.3, 9.5 in your textbook. Tuesday (4/28) Read and work through questions 1-9 Wednesday (4/29) Read and work through questions 10-14 Thursday (4/30) Read and work through questions 14-18 Friday (5/31) Read and work through questions 19-21 How are the chemical formula and name of a molecular compound related? Why? When you began chemistry class this year, you probably already knew that the chemical formula for carbon dioxide was CO2. Today you will find out why CO2 is named that way. Naming chemical compounds correctly is of paramount importance. The slight difference between the names carbon monoxide (CO, a poisonous, deadly gas) and carbon dioxide (CO2, a greenhouse gas that we exhale when we breathe out) can be the difference between life and death! In this activity you will learn the naming system for molecular compounds. Model 1 – Molecular Compounds Molecular Number of Atoms Number of Atoms in Name of Compound Formula of First Element Second Element ClF Chlorine monofluoride ClF5 1 5 Chlorine pentafluoride CO Carbon monoxide CO2 Carbon dioxide Cl2O Dichlorine monoxide PCl5 Phosphorus pentachloride N2O5 Dinitrogen pentoxide 1. Fill in the table to indicate the number of atoms of each type in the molecular formula. -

CDG Solution 3000TM Safety Data Sheet
Emergency Telephone Number: 800-424-9300 24 hours/day, 7 days/week Emergency Telephone Number: 800-424-9300 24 hours, 7 days/week CDG Solution 3000TM Safety Data Sheet Section 1 – Chemical Product and Company Identification Product Identifier: Chlorine Dioxide 0.3% Aqueous Solution SDS No. CD-004 Other Means of Identification: Chlorine Oxide Solution Chlorine Peroxide Solution Chlorine (IV) Oxide Solution Chloroperoxyl Solution Recommended Use: Biocide Company: CDG Environmental, LLC 301 Broadway, Suite 420 Bethlehem, PA 18015 888-610-2562 484-821-0780 Emergency Phone Number: 800-424-9300 SDS # CD-004 CDG Solution 3000 Page 1 of 12 Emergency Telephone Number: 800-424-9300 24 hours, 7 days/week Section 2 – Hazards Identification GHS Classification: Skin Irritation: Category 2 Eye Irritation: Category 2B Acute Toxicity – Inhalation: Category 4 Signal Word: Warning Pictogram: Hazard Statements: Causes skin irritation Causes eye irritation Harmful in inhaled Precautionary Wash exposed areas thoroughly after handling. Statements: Wear protective gloves. Avoid breathing fume/gas/mist/vapors/spray. Use only outdoors or in a well-ventilated area. If on skin: Wash with plenty of water. If skin irritation occurs: Get medical attention. If in eyes: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. If eye irritation persists: Get medical attention. If inhaled: Remove person to fresh air and keep comfortable for breathing. Call a doctor if you feel unwell. Specific treatment (see First -

Toxicological Profile for Chlorine Dioxide and Chlorite
TOXICOLOGICAL PROFILE FOR CHLORINE DIOXIDE AND CHLORITE U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Agency for Toxic Substances and Disease Registry September 2004 CHLORINE DIOXIDE AND CHLORITE ii DISCLAIMER The use of company or product name(s) is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry. CHLORINE DIOXIDE AND CHLORITE iii UPDATE STATEMENT Toxicological Profile for Chlorine Dioxide and Chlorite, Draft for Public Comment was released in September 2002. This edition supersedes any previously released draft or final profile. Toxicological profiles are revised and republished as necessary. For information regarding the update status of previously released profiles, contact ATSDR at: Agency for Toxic Substances and Disease Registry Division of Toxicology/Toxicology Information Branch 1600 Clifton Road NE, Mailstop F-32 Atlanta, Georgia 30333 CHLORINE DIOXIDE AND CHLORITE vi *Legislative Background The toxicological profiles are developed in response to the Superfund Amendments and Reauthorization Act (SARA) of 1986 (Public law 99-499) which amended the Comprehensive Environmental Response, Compensation, and Liability Act of 1980 (CERCLA or Superfund). This public law directed ATSDR to prepare toxicological profiles for hazardous substances most commonly found at facilities on the CERCLA National Priorities List and that pose the most significant potential threat to human health, as determined by ATSDR and the EPA. The availability of the revised priority list of 275 hazardous substances was announced in the Federal Register on November 17, 1997 (62 FR 61332). For prior versions of the list of substances, see Federal Register notices dated April 29, 1996 (61 FR 18744); April 17, 1987 (52 FR 12866); October 20, 1988 (53 FR 41280); October 26, 1989 (54 FR 43619); October 17, 1990 (55 FR 42067); October 17, 1991 (56 FR 52166); October 28, 1992 (57 FR 48801); and February 28, 1994 (59 FR 9486). -

Heterogeneous Production of Perchlorate and Chlorate By
HETEROGENEOUS PRODUCTION OF PERCHLORATE AND CHLORATE BY OZONE OXIDATION OF Cl- by SIXUAN WANG B.E. A Thesis In CIVIL ENGINEERING Submitted to the Graduate Faculty of Texas Tech University in Partial Fulfillment of The Requirements for The Degree of MASTER OF SCIENCE IN CIVIL ENGINEERING Approved Dr. Andrew W. Jackson Chairperson of the Committee Dr. Todd A. Anderson Peggy Gordon Dean of Graduate School August, 2011 Copyright © 2011, Sixuan Wang Texas Tech University, Sixuan Wang, August 2011 ACKNOWLEDGEMENTS This research project would not finish without the support of many people. I would like to express my gratitude to all those who gave me help and support to complete this thesis. I want to thank Dr. Andrew Jackson, the chair of my committee for giving me the chance, guidance, and encouragement. I would like to take this opportunity to thank my committee members, Dr. Todd Anderson, for his continuous support and encouragement all the way. I would also take this opportunity to thank my friends and colleagues Dr. Rao, Nubia, Felipe, Tony and Tim for their continuous help, sharing during these two years of research in Environmental lab. I am very thankful to the Strategic Environmental Research and Development Program (SERDP) for providing the necessary funds for this project. ii Texas Tech University, Sixuan Wang, August 2011 TABLE OF CONTENTS ACKNOWLEDGEMENTS .......................................................................................... ii ABSTRACT ................................................................................................................. -

Example Exercise 8.1 Evidence for a Reaction
Example Exercise 8.1 Evidence for a Reaction Which of the following is experimental evidence for a chemical reaction? (a) Pouring vinegar on baking soda gives foamy bubbles. (b) Mixing two solutions produces insoluble particles. (c) Mixing two colorless solutions gives a yellow solution. (d) Mixing two solutions produces a temperature increase. Solution We can analyze each of these observations based on the criteria for a chemical reaction. (a) The bubbles produced indicate a chemical reaction is occurring. (b) The insoluble particles formed indicate a chemical reaction. (c) The yellow color produced indicates a chemical reaction. (d) The temperature increase indicates heat energy is being released and, thus, an exothermic chemical reaction. Practice Exercise What are four observations that a chemical reaction has occurred? Answers: (a) a gas is released; (b) a precipitate is produced; (c) a permanent color change is observed; (d) an energy change is noted Concept Exercise What four observations are evidence for a chemical reaction? Answer: See Appendix G. Introductory Chemistry: Concepts and Critical Thinking, 6th Edition © 2011 Pearson Education, Inc. Charles H. Corwin Example Exercise 8.2 Writing Chemical Equations Write a chemical equation for each of the following chemical reactions: (a) Mercury liquid and fluorine gas react to give solid mercury(II) fluoride. (b) Zinc metal reacts with sulfuric acid to give aqueous zinc sulfate and hydrogen gas. Solution To write the chemical equation, we must provide formulas and symbols for each substance. We can describe each of the preceding chemical reactions as follows: (a) Hg(l) + F2(g) → HgF2(s) (b) Zn(s) + H2SO4(aq) → ZnSO4(aq) + H2(g) Practice Exercise Write a chemical equation for each of the following chemical reactions: (a) Aqueous solutions of sodium iodide and silver nitrate yield silver iodide precipitate and aqueous sodium nitrate. -

18 Rapid Disinfection of Combined Sewer Overflow Using Chlorine Dioxide
18 Rapid Disinfection of Combined Sewer Overflow Using Chlorine Dioxide R. Takahashi1 , T.Kirihara2 , M.Koeda3 1 Director, 2 Chief Researcher, 3 Senior Researcher Japan Institute of Wastewater Engineering Technology ABSTRACT This is a technology in which disinfection of untreated sewage from pump stations in combined sewer systems and primary effluent from final treatment plants is performed by oxidization using chlorine dioxide. As a distinctive feature of the technology, the disinfectant is rapidly and uniformly injected and diffused in the water channel cross-section, resulting in substantially improved disinfection efficiency in comparison with conventional methods using chlorine disinfectants such as sodium chlorite. KEYWORDS Disinfection, chlorine dioxide, ClO2 generation system, ClO2 injection system 1. OUTLINE OF TECHNOLOGY 1.1 Principle of technology Chlorine dioxide is expressed by the chemical formula ClO2, and is a reddish-yellow gas with a pungent odor resembling chlorine or ozone at room temperature. Its physical constants are shown in Table 1. Chlorine oxide is water soluble and exists in aqueous solutions in the form of ClO2. Because it is strongly oxidizing and has the following distinctive properties, it is considered suitable for use as a disinfectant in combined sewers: 1) Rapid disinfection effect in comparison with chlorine agents (sodium hypochlorite, etc.), 2) In the range of pH 6-10, the effect of pH on disinfection effectiveness is not as great as with chlorine, 3) Does not react with ammonia, 4) Does not form tri-halide-methane, and 5) Does not display persistence, and therefore has little adverse effect on public waters. Furthermore, contact with coliform groups is achieved within a short time as a result of rapid and uniform injection/diffusion of ClO2 in the water channel cross-section. -

Chlorine Dioxide Incident Management
Chlorine Dioxide Incident Management Key Points Fire not combustible, but enhances combustion of other substances oxidising agent reacts violently with organics, phosphorus, potassium hydroxide and sulphur, causing a fire and explosion hazard emits toxic fumes of chlorine when heated to decomposition; reacts with water to produce hydrochloric acid and chloric acid Health major routes of exposure are inhalation or ocular inhalation causes irritation of eyes and nose with sore throat, cough, chest tightness, headache, fever, wheeze, tachycardia and confusion chemical pneumonitis, tachypnoea, dyspnoea and stridor due to laryngeal oedema may follow inhalation exposure corrosive damage to respiratory tract occurs in serious cases; severe injuries may result in persistent hoarseness, pulmonary fibrosis and chronic obstructive airway disease Environment hazardous to the environment; inform the Environment Agency of substantial incidents PHE publications gateway number: 2014790 Published: January 2016 Compendium of Chemical Hazards: Chlorine Dioxide Hazard Identification Standard (UK) dangerous goods emergency action codes Data not available Classification, labelling and packaging (CLP)* Chlorine dioxide (in aqueous solution) Hazard class and Acute Tox. 3 Acute toxicity (oral), category 3 category Skin Corr. 1B Skin corrosion, category 1B Aquatic Acute 1 Acute hazards to the aquatic environment, category 1 Hazard statement H301 Toxic if swallowed H314 Causes severe skin burns and eye damage H400 Very toxic to aquatic life Signal words DANGER * Implemented in the EU on 20 January 2009 Reference European Commission. Harmonised classification – Annexe VI to Regulation (EC) No. 1272/2008 on Classification, Labelling and Packaging of Substances and Mixtures. http://echa.europa.eu/information-on-chemicals/cl-inventory- database (accessed 07/2015). -

Theoretical Photoabsorption Spectra of Cloocl and Cl2o
7278 J. Phys. Chem. A 2000, 104, 7278-7283 Theoretical Photoabsorption Spectra of ClOOCl and Cl2O A. Toniolo,† M. Persico,* and D. Pitea† Dipartimento di Chimica Fisica ed Elettrochimica, UniVersita` di Milano, Via Golgi 19, I-20133 Milano, Italy, and Dipartimento di Chimica e Chimica Industriale, UniVersita` di Pisa, Via Risorgimento 35, I-56126 Pisa, Italy ReceiVed: March 27, 2000; In Final Form: May 25, 2000 We calculated transition energies and oscillator strengths of ClOOCl and Cl2O in the Franck-Condon region with ab initio techniques. We describe 8 singlet excited states for ClOOCl and 13 excited states for Cl2O. The photoabsorption spectra are simulated by a semiclassical approach, which takes into account the vibrational phase space distribution in the ground state and the geometry dependence of excitation energies and transition dipoles. We present an unambiguous assignation of the available UV spectra, which is preliminary to the interpretation of the photochemistry of these compounds. 1. Introduction In the last fifteen years, the photochemistry of chlorine oxides has received considerable attention: the importance of the ozone loss catalytic cycles has made advisable to understand the photodissociation reactions of the chlorine-oxygen containing molecules present in the stratosphere.1-3 Among the important chlorine-oxygen compounds, the spectroscopy of ClO has been investigated theoretically by us and others,4-6 and rather complete potential energy surfaces for 7 ClO2 have been presented. ClOOCl and Cl2O have been paid less attention by computational chemists. It is now accepted that the most important catalytic cycle that results in ozone depletion in the Antarctic stratosphere starts with the self-reaction of ClO radicals and involves the formation of a chlorine peroxide ClOOCl molecule.8 The absorption of radiation breaks preferably one of the two Cl-O Figure 1. -

Dichlorine Monoxide
Dichlorine monoxide Other names: Chlorine monooxide; Chlorine monoxide; Chlorine monoxide (Cl2O); Chlorine oxide; Chlorine oxide (Cl2O); Cl2O; Dichlorine oxide; Dichloromonoxide; Dichloroxide; Hypochlorous anhydride. InChI: InChI=1S/Cl2O/c1-3-2 InChI Key: RCJVRSBWZCNNQT-UHFFFAOYSA-N Formula: Cl2O SMILES: ClOCl Molecular Weight: 86.90 CAS: 7791-21-1 Physical Properties Property Value Unit Source EA 1.30 ± 0.43 eV NIST Webbook ∆ G° -179.74 Joback Method f kJ/mol ∆ H° -207.03 Joback Method f gas kJ/mol ∆ H° 5.34 Joback Method fus kJ/mol ∆ H° 26.77 Joback Method vap kJ/mol IE 10.91 ± 0.02 eV NIST Webbook IE 10.91 ± 0.02 eV NIST Webbook IE 10.94 eV NIST Webbook IE 11.20 ± 0.10 eV NIST Webbook IE 11.02 eV NIST Webbook logP 1.31 Crippen Method oct/wat P 5836.07 Joback Method c kPa T 296.68 Joback Method boil K T 479.25 Joback Method c K T 171.83 Joback Method fus K V 0.15 3 Joback Method c m /kg-mol Temperature Dependent Properties Property Value Unit Temperature (K) Source C 47.60 J/mol×K 296.68 Joback Method p,gas η 0.00 Pa×s 296.68 Joback Method Sources Joback Method: https://en.wikipedia.org/wiki/Joback_method NIST Webbook: http://webbook.nist.gov/cgi/inchi/InChI=1S/Cl2O/c1-3-2 Crippen Method: http://pubs.acs.org/doi/abs/10.1021/ci990307l Legend C : Ideal gas heat capacity (J/mol×K). p,gas η: Dynamic viscosity (Pa×s). EA: Electron affinity (eV). ∆ G°: Standard Gibbs free energy of formation (kJ/mol). -
![Appendix 3-3 Ignition / Explosion Risk Caused by Blending [Safety Guidelines for Chemical Experiments, 4Th Edition] by the Chemical Society of Japan, Maruzen (1999)](https://docslib.b-cdn.net/cover/1640/appendix-3-3-ignition-explosion-risk-caused-by-blending-safety-guidelines-for-chemical-experiments-4th-edition-by-the-chemical-society-of-japan-maruzen-1999-3001640.webp)
Appendix 3-3 Ignition / Explosion Risk Caused by Blending [Safety Guidelines for Chemical Experiments, 4Th Edition] by the Chemical Society of Japan, Maruzen (1999)
Supplement 1. Safe Handling of Hazardous Substances Appendix 3-3 Ignition / Explosion Risk Caused by Blending [Safety Guidelines for Chemical Experiments, 4th Edition] by the Chemical Society of Japan, Maruzen (1999) This table has been classified and structured according to the chemical structure based on the NFPA’s (National Fire Protection Association) “Manual of Hazardous Chemical Reaction 491M (1975),” in which examples of accident cases and hazardous reaction cases involving chemical substances blending have been compiled. 1. Oxidizing Substance & Flammable Substance 1) Oxidizing Substance 2) Flammable Substance a) oxo-halogen acid salt a) non-metal elemental substance perchlorate, chlorate, bromate, iodate, chlorite, phosphorus, sulfur, activated carbon, etc. hypochlorite, etc. b) metallic peroxide, hydrogen peroxide b) metal metallic peroxide: potassium peroxide, calcium magnesium, zinc, aluminum, etc. peroxide, etc. c) permanganate c) sulfide potassium permanganate, etc. phosphorus sulfide, antimony sulfide, carbon bisulfide, etc. d) nichrome acid salt d) hydride dichromate potassium, etc. silane, phosphine, dibrane, arsine, etc. e) nitrate salt e) carbide potassium nitrate, sodium nitrate, ammonium calcium carbide nitrate, etc. f) nitric acid, fuming nitric acid f) organic substance hydrocarbon, alcohol, ketone, organic acid, amine, etc. g) nitric acid, fuming nitric acid, chlorosulfuric g) other acid metal amid, cyanide, hydroxylamine, etc. h) chrome oxide (III) i) perchloric acid j) peroxodisulfuric acid k) chlorine oxide, chlorine dioxide, chlorine monoxide l) nitrogen dioxide (nitrogen tetroxide) m) halogen fluorine, chlorine, bromine, iodine, chlorine trifluoride, bromine trifluoride, iodine trifluoride, chlorine pentafluoride, bromine pentafluoride, iodine pentafluoride n) halogenation nitrogen nitrogen trifluoride, nitrogen trichloride, nitrogen tribromide, nitrogen triiodide 2. Hydrogen Peroxide & Metallic Oxide 3. Persulfate & Manganese metallic oxide managanese dioxide, mercuric oxide 4. -

Chapter 4 Nomenclature
Chapter 4 Nomenclature 1. A binary compound is one that contains only two elements. Examples are sodium chloride, water, and carbon dioxide. 2. compounds that contain a metal and a nonmetal; compounds containing two nonmetals 3. cation, anion 4. cation 5. The substance “sodium chloride” consists of an extended lattice array of sodium ions, Na+, and chloride ions, Cl–. Each sodium ion is surrounded by several chloride ions, and each chloride ion is surrounded by several sodium ions. We write the formula as NaCl to indicate the relative number of each ion in the substance. 6. a. sodium iodide b. calcium fluoride c. aluminum sulfide d. calcium bromide e. strontium oxide f. silver chloride [silver(I) chloride] g. cesium iodide h. lithium oxide 7. a. Incorrect; BaH2 is barium hydride. b. Incorrect; Na2O is sodium oxide. c. Correct d. Incorrect; SiO2 is silicon dioxide. e. Correct 8. a. Since each iodide ion has a 1– charge, the iron ion must have a 3+ charge: the name is iron(III) iodide. b. Since each chloride ion has a 1– charge, the manganese must have a 2+ charge: the name is manganese(II) chloride. c. Since the oxide ion has a 2– charge, the mercury ion must have a 2+ charge: the name is mercury(II) oxide. d. Since the oxide ion has a 2– charge, the copper atoms must each have a 1+ charge: the name is copper(I) oxide. e. Since the oxide ion has a 2– charge, the copper ion must have a 2+ charge: copper(II) oxide. f. -

Chlorine Dioxide
Committee on Acute Exposure Guideline Levels Committee on Toxicology Board on Environmental Studies and Toxicology Division on Earth and Life Studies THE NATIONAL ACADEMIES PRESS 500 Fifth Street, NW Washington, DC 20001 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Insti- tute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for ap¬propriate balance. This project was supported by Contract No. DAMD17-99-C-9049 between the National Academy of Sciences and the U.S. Department of Defense and Contract No. 68-C-03-081 between the National Academy of Sciences and the U.S. Environmental Protection Agency. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the or- ganizations or agencies that provided support for this project. International Standard Book Number-13: 978-0-309-10358-9 International Standard Book Number-10: 0-309-10358-4 Additional copies of this report are available from The National Academies Press 500 Fifth Street, NW Box 285 Washington, DC 20055 800-624-6242 202-334-3313 (in the Washington metropolitan area) http://www.nap.edu Copyright 2007 by the National Academy of Sciences. All rights reserved. Printed in the United States of America The National Academy of Sciences is a private, nonprofit, self-perpetuating society of distinguished scholars engaged in scientific and engineering research, dedicated to the furtherance of science and technology and to their use for the general welfare.