The TB Structural Genomics Consortium: a Resource for Mycobacterium Tuberculosis Biology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lignite Coal Burning Seam in the Remote Altai Mountains Harbors A
www.nature.com/scientificreports OPEN Lignite coal burning seam in the remote Altai Mountains harbors a hydrogen-driven thermophilic Received: 20 September 2017 Accepted: 17 April 2018 microbial community Published: xx xx xxxx Vitaly V. Kadnikov1, Andrey V. Mardanov1, Denis A. Ivasenko2, Dmitry V. Antsiferov2, Alexey V. Beletsky1, Olga V. Karnachuk2 & Nikolay V. Ravin1 Thermal ecosystems associated with underground coal combustion sites are rare and less studied than geothermal features. Here we analysed microbial communities of near-surface ground layer and bituminous substance in an open quarry heated by subsurface coal fre by metagenomic DNA sequencing. Taxonomic classifcation revealed dominance of only a few groups of Firmicutes. Near- complete genomes of three most abundant species, ‘Candidatus Carbobacillus altaicus’ AL32, Brockia lithotrophica AL31, and Hydrogenibacillus schlegelii AL33, were assembled. According to the genomic data, Ca. Carbobacillus altaicus AL32 is an aerobic heterotroph, while B. lithotrophica AL31 is a chemolithotrophic anaerobe assimilating CO2 via the Calvin cycle. H. schlegelii AL33 is an aerobe capable of both growth on organic compounds and carrying out CO2 fxation via the Calvin cycle. Phylogenetic analysis of the large subunit of RuBisCO of B. lithotrophica AL31 and H. schlegelii AL33 showed that it belongs to the type 1-E. All three Firmicutes species can gain energy from aerobic or anaerobic oxidation of molecular hydrogen, produced as a result of underground coal combustion along with other coal gases. We propose that thermophilic Firmicutes, whose spores can spread from their original geothermal habitats over long distances, are the frst colonizers of this recently formed thermal ecosystem. Studies of thermophilic microorganisms that survive and develop at temperatures that are extreme for ordi- nary life have broadened our understanding of the diversity of microorganisms and their evolution, the mech- anisms of adaptation to environmental conditions. -

Supporting Information High-Throughput Virtual Screening
Supporting Information High-Throughput Virtual Screening of Proteins using GRID Molecular Interaction Fields Simone Sciabola, Robert V. Stanton, James E. Mills, Maria M. Flocco, Massimo Baroni, Gabriele Cruciani, Francesca Perruccio and Jonathan S. Mason Contents Table S1 S2-S21 Figure S1 S22 * To whom correspondence should be addressed: Simone Sciabola, Pfizer Research Technology Center, Cambridge, 02139 MA, USA Phone: +1-617-551-3327; Fax: +1-617-551-3117; E-mail: [email protected] S1 Table S1. Description of the 990 proteins used as decoy for the Protein Virtual Screening analysis. PDB ID Protein family Molecule Res. (Å) 1n24 ISOMERASE (+)-BORNYL DIPHOSPHATE SYNTHASE 2.3 1g4h HYDROLASE 1,3,4,6-TETRACHLORO-1,4-CYCLOHEXADIENE HYDROLASE 1.8 1cel HYDROLASE(O-GLYCOSYL) 1,4-BETA-D-GLUCAN CELLOBIOHYDROLASE I 1.8 1vyf TRANSPORT PROTEIN 14 KDA FATTY ACID BINDING PROTEIN 1.85 1o9f PROTEIN-BINDING 14-3-3-LIKE PROTEIN C 2.7 1t1s OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 2.4 1t1r OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 2.3 1q0q OXIDOREDUCTASE 1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE 1.9 1jcy LYASE 2-DEHYDRO-3-DEOXYPHOSPHOOCTONATE ALDOLASE 1.9 1fww LYASE 2-DEHYDRO-3-DEOXYPHOSPHOOCTONATE ALDOLASE 1.85 1uk7 HYDROLASE 2-HYDROXY-6-OXO-7-METHYLOCTA-2,4-DIENOATE 1.7 1v11 OXIDOREDUCTASE 2-OXOISOVALERATE DEHYDROGENASE ALPHA SUBUNIT 1.95 1x7w OXIDOREDUCTASE 2-OXOISOVALERATE DEHYDROGENASE ALPHA SUBUNIT 1.73 1d0l TRANSFERASE 35KD SOLUBLE LYTIC TRANSGLYCOSYLASE 1.97 2bt4 LYASE 3-DEHYDROQUINATE DEHYDRATASE -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Polymorphic Positions in the Genome of Xcm Page 1 of 5
Polymorphic positions in the genome of Xcm seq_id position 2005 2251 4387 4389 4379 4380 4381 4383 4384 4394 4392 4395 4433 4434 genes silent/non-silent NZ_ACHT01000023 16591 G t t t t t t t t t t t t t Intergenic Intergenic NZ_ACHT01000028 7775 A c c c c c c c c c c c c c XcampmN_010100000587 ribosomal protein alanine acetyltransferase (7614-8207) non-silent tac -> taA; NZ_ACHT01000041 15615 C t t t t t t t t t t t t t XcampmN_010100000977 hemolysin III (15149-15805) non-silent atc -> aCc; NZ_ACHT01000044 2934 T c c c c c c c c c c c c c XcampmN_010100001152 hypothetical protein (2671-3549) non-silent gat -> Aat; NZ_ACHT01000044 3736 A g g g g g g g g g g g g g XcampmN_010100001157 bifunctional aspartate kinase/diaminopimelate decarboxylase non-silentprotein (3730-4207) gct -> Act; NZ_ACHT01000060 1984 C a a a a a a a a a a a a a Intergenic Intergenic NZ_ACHT01000064 1939 C t t t t t t t t t t t t t Intergenic Intergenic NZ_ACHT01000072 4507 A c c c c c c c c c c c c c XcampmN_010100002109 VirB3 protein (4240-4551) non-silent atg -> atT; NZ_ACHT01000089 30693 A c c c c c c c c c c c c c XcampmN_010100002947 hypothetical protein (30250-31116) silent atc -> atA; NZ_ACHT01000101 1035 T c c c c c c c c c c c c c XcampmN_010100003517 soluble lytic murein transglycosylase (1-1051) non-silent cgc -> cAc; NZ_ACHT01000101 2893 T c c c c c c c c c c c c c XcampmN_010100003522 TonB-dependent receptor (1308-4187) non-silent gcg -> gTg; NZ_ACHT01000120 419 A c c c c c c c c c c c c c XcampmN_010100004174 acyl carrier protein phosphodiesterase (162-746) -

Suppl Fürcorel Ohne Mend Ohnetextfelder.Cdr
Additional File 1 Fermentation stage-dependent adaptations of Bacillus licheniformis during enzyme production Sandra Wiegand, Birgit Voigt, Dirk Albrecht, Johannes Bongaerts, Stefan Evers, Michael Hecker, Rolf Daniel and Heiko Liesegang Content Figure S1 Protease production and process parameters. ........................................................................... 2 Figure S2 Proteome of the amino acid metabolism – Part I. .................................................................... 3 Figure S3 Proteome of the amino acid metabolism – Part II. ................................................................... 4 Figure S4 Amino acid transport. ............................................................................................................... 5 Figure S5 Proteome of the central carbon metabolism. ............................................................................ 7 Figure S6 Carbohydrate transport. ............................................................................................................ 8 Figure S7 Acetoin utilization operon acuABC. ....................................................................................... 10 Figure S8 Most abundant proteins. ......................................................................................................... 10 Figure S9 Lichenicidin gene cluster. ....................................................................................................... 11 Figure S10 Cell envelope stress response. ............................................................................................. -

Supplementary Material
Supplementary material Figure S1. Cluster analysis of the proteome profile based on qualitative data in low and high sugar conditions. Figure S2. Expression pattern of proteins under high and low sugar cultivation of Granulicella sp. WH15 a) All proteins identified in at least two out of three replicates (excluding on/off proteins). b) Only proteins with significant change t-test p=0.01. 2fold change is indicated by a red line. Figure S3. TigrFam roles of the differentially expressed proteins, excluding proteins with unknown function. Figure S4. General overview of up (red) and downregulated (blue) metabolic pathways based on KEGG analysis of proteome. Table S1. growth of strain Granulicella sp. WH15 in culture media supplemented with different carbon sources. Carbon Source Growth Pectin - Glycogen - Glucosamine - Cellulose - D-glucose + D-galactose + D-mannose + D-xylose + L-arabinose + L-rhamnose + D-galacturonic acid - Cellobiose + D-lactose + Sucrose + +=positive growth; -=No growth. Table S2. Total number of transcripts reads per sample in low and high sugar conditions. Sample ID Total Number of Reads Low sugar (1) 15,731,147 Low sugar (2) 12,624,878 Low sugar (3) 11,080,985 High sugar (1) 11,138,128 High sugar (2) 9,322,795 High sugar (3) 10,071,593 Table S3. Differentially up and down regulated transcripts in high sugar treatment. ORF Annotation Log2FC GWH15_14040 hypothetical protein 3.71 GWH15_06005 hypothetical protein 3.12 GWH15_00285 tRNA-Asn(gtt) 2.74 GWH15_06010 hypothetical protein 2.70 GWH15_14055 hypothetical protein 2.66 -
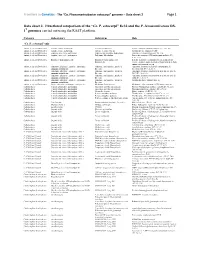
Ca. P. Ectocarpi” Ec32 and the P
Frontiers in Genetics - The “Ca. Phaeomarinobacter ectocarpi” genome – Data sheet 2 Page 1 Data sheet 2. Functional comparison of the “Ca. P. ectocarpi” Ec32 and the P. lavamentivorans DS- 1T genomes carried out using the RAST platform. Category Subcategory Subsystem Role “Ca. P. ectocarpi” only Amino Acids and Derivatives Alanine, serine, and glycine Alanine biosynthesis Valine--pyruvate aminotransferase (EC 2.6.1.66) Amino Acids and Derivatives Alanine, serine, and glycine Glycine cleavage system Sodium/glycine symporter GlyP Amino Acids and Derivatives Arginine; urea cycle, polyamines Arginine and Ornithine Degradation Ornithine cyclodeaminase (EC 4.3.1.12) Amino Acids and Derivatives Arginine; urea cycle, polyamines Polyamine Metabolism Putrescine transport ATP-binding protein PotA (TC 3.A.1.11.1) Amino Acids and Derivatives Branched-chain amino acids Branched-Chain Amino Acid Leucine-responsive regulatory protein, regulator for Biosynthesis leucine (or lrp) regulon and high-affinity branched-chain amino acid transport system Amino Acids and Derivatives Glutamine, glutamate, aspartate, asparagine; Glutamate and Aspartate uptake in Glutamate Aspartate periplasmic binding protein ammonia assimilation Bacteria precursor GltI (TC 3.A.1.3.4) Amino Acids and Derivatives Glutamine, glutamate, aspartate, asparagine; Glutamate and Aspartate uptake in Glutamate Aspartate transport system permease protein ammonia assimilation Bacteria GltJ (TC 3.A.1.3.4) Amino Acids and Derivatives Glutamine, glutamate, aspartate, asparagine; Glutamate and Aspartate -

Supplemental Table S1: Comparison of the Deleted Genes in the Genome-Reduced Strains
Supplemental Table S1: Comparison of the deleted genes in the genome-reduced strains Legend 1 Locus tag according to the reference genome sequence of B. subtilis 168 (NC_000964) Genes highlighted in blue have been deleted from the respective strains Genes highlighted in green have been inserted into the indicated strain, they are present in all following strains Regions highlighted in red could not be deleted as a unit Regions highlighted in orange were not deleted in the genome-reduced strains since their deletion resulted in severe growth defects Gene BSU_number 1 Function ∆6 IIG-Bs27-47-24 PG10 PS38 dnaA BSU00010 replication initiation protein dnaN BSU00020 DNA polymerase III (beta subunit), beta clamp yaaA BSU00030 unknown recF BSU00040 repair, recombination remB BSU00050 involved in the activation of biofilm matrix biosynthetic operons gyrB BSU00060 DNA-Gyrase (subunit B) gyrA BSU00070 DNA-Gyrase (subunit A) rrnO-16S- trnO-Ala- trnO-Ile- rrnO-23S- rrnO-5S yaaC BSU00080 unknown guaB BSU00090 IMP dehydrogenase dacA BSU00100 penicillin-binding protein 5*, D-alanyl-D-alanine carboxypeptidase pdxS BSU00110 pyridoxal-5'-phosphate synthase (synthase domain) pdxT BSU00120 pyridoxal-5'-phosphate synthase (glutaminase domain) serS BSU00130 seryl-tRNA-synthetase trnSL-Ser1 dck BSU00140 deoxyadenosin/deoxycytidine kinase dgk BSU00150 deoxyguanosine kinase yaaH BSU00160 general stress protein, survival of ethanol stress, SafA-dependent spore coat yaaI BSU00170 general stress protein, similar to isochorismatase yaaJ BSU00180 tRNA specific adenosine -

Methylcitrate Cycle Defines the Bactericidal Essentiality of Isocitrate Lyase for Survival of Mycobacterium Tuberculosis on Fatty Acids
Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of Mycobacterium tuberculosis on fatty acids Hyungjin Eoh and Kyu Y. Rhee1 Department of Medicine, Division of Infectious Diseases, Weill Cornell Medical College, New York, NY 10065 Edited by William R. Jacobs, Jr., Albert Einstein College of Medicine of Yeshiva University, Bronx, NY, and approved February 25, 2014 (received for review January 8, 2014) Few mutations attenuate Mycobacterium tuberculosis (Mtb) more tracing to elucidate the intrabacterial metabolic fates of an even profoundly than deletion of its isocitrate lyases (ICLs). However, (acetate) and odd (propionate) chain fatty acid in ICL-deficient the basis for this attenuation remains incompletely defined. Mtb’s Mtb and identify a unifying biochemical mechanism that explains its ICLs are catalytically bifunctional isocitrate and methylisocitrate vulnerability to both even- and odd-chain fatty acids. lyases required for growth on even and odd chain fatty acids. Here, we report that Mtb’s ICLs are essential for survival on both Results acetate and propionate because of its methylisocitrate lyase (MCL) Bactericidal Activity of Acetate and Propionate Against ICL-Deficient activity. Lack of MCL activity converts Mtb’s methylcitrate cycle Mtb. Previous studies reported that, consistent with predicted into a “dead end” pathway that sequesters tricarboxylic acid defects in the glyoxylate shunt and methylcitrate cycles, ICL- (TCA) cycle intermediates into methylcitrate cycle intermediates, deficient Mtb were specifically unable to grow when cultured in depletes gluconeogenic precursors, and results in defects of mem- media containing the model fatty acid metabolites, acetate or brane potential and intrabacterial pH. Activation of an alternative propionate (4–6). -

Demystifying the Catalytic Pathway of Mycobacterium Tuberculosis Isocitrate Lyase Collins U
www.nature.com/scientificreports OPEN Demystifying the catalytic pathway of Mycobacterium tuberculosis isocitrate lyase Collins U. Ibeji1,2,3, Nor Amirah Mohd Salleh1, Jia Siang Sum1, Angela Chiew Wen Ch’ng1, Theam Soon Lim1 & Yee Siew Choong1* Pulmonary tuberculosis, caused by Mycobacterium tuberculosis, is one of the most persistent diseases leading to death in humans. As one of the key targets during the latent/dormant stage of M. tuberculosis, isocitrate lyase (ICL) has been a subject of interest for new tuberculosis therapeutics. In this work, the cleavage of the isocitrate by M. tuberculosis ICL was studied using quantum mechanics/ molecular mechanics method at M06-2X/6-31+G(d,p): AMBER level of theory. The electronic embedding approach was applied to provide a better depiction of electrostatic interactions between MM and QM regions. Two possible pathways (pathway I that involves Asp108 and pathway II that involves Glu182) that could lead to the metabolism of isocitrate was studied in this study. The results suggested that the core residues involved in isocitrate catalytic cleavage mechanism are Asp108, Cys191 and Arg228. A water molecule bonded to Mg2+ acts as the catalytic base for the deprotonation of isocitrate C(2)–OH group, while Cys191 acts as the catalytic acid. Our observation suggests that the shuttle proton from isocitrate hydroxyl group C(2) atom is favourably transferred to Asp108 instead of Glu182 with a lower activation energy of 6.2 kcal/mol. Natural bond analysis also demonstrated that pathway I involving the transfer of proton to Asp108 has a higher intermolecular interaction and charge transfer that were associated with higher stabilization energy. -

Genome-Scale Metabolic Network Analysis and Drug Targeting of Multi-Drug Resistant Pathogen Acinetobacter Baumannii AYE
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2017 Electronic Supplementary Information (ESI) for Molecular BioSystems Genome-scale metabolic network analysis and drug targeting of multi-drug resistant pathogen Acinetobacter baumannii AYE Hyun Uk Kim, Tae Yong Kim and Sang Yup Lee* E-mail: [email protected] Supplementary Table 1. Metabolic reactions of AbyMBEL891 with information on their genes and enzymes. Supplementary Table 2. Metabolites participating in reactions of AbyMBEL891. Supplementary Table 3. Biomass composition of Acinetobacter baumannii. Supplementary Table 4. List of 246 essential reactions predicted under minimal medium with succinate as a sole carbon source. Supplementary Table 5. List of 681 reactions considered for comparison of their essentiality in AbyMBEL891 with those from Acinetobacter baylyi ADP1. Supplementary Table 6. List of 162 essential reactions predicted under arbitrary complex medium. Supplementary Table 7. List of 211 essential metabolites predicted under arbitrary complex medium. AbyMBEL891.sbml Genome-scale metabolic model of Acinetobacter baumannii AYE, AbyMBEL891, is available as a separate file in the format of Systems Biology Markup Language (SBML) version 2. Supplementary Table 1. Metabolic reactions of AbyMBEL891 with information on their genes and enzymes. Highlighed (yellow) reactions indicate that they are not assigned with genes. No. Metabolism EC Number ORF Reaction Enzyme R001 Glycolysis/ Gluconeogenesis 5.1.3.3 ABAYE2829 -

Methylcitrate Cycle Defines the Bactericidal Essentiality of Isocitrate Lyase for Survival of Mycobacterium Tuberculosis on Fatty Acids
Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of Mycobacterium tuberculosis on fatty acids Hyungjin Eoh and Kyu Y. Rhee1 Department of Medicine, Division of Infectious Diseases, Weill Cornell Medical College, New York, NY 10065 Edited by William R. Jacobs, Jr., Albert Einstein College of Medicine of Yeshiva University, Bronx, NY, and approved February 25, 2014 (received for review January 8, 2014) Few mutations attenuate Mycobacterium tuberculosis (Mtb) more tracing to elucidate the intrabacterial metabolic fates of an even profoundly than deletion of its isocitrate lyases (ICLs). However, (acetate) and odd (propionate) chain fatty acid in ICL-deficient the basis for this attenuation remains incompletely defined. Mtb’s Mtb and identify a unifying biochemical mechanism that explains its ICLs are catalytically bifunctional isocitrate and methylisocitrate vulnerability to both even- and odd-chain fatty acids. lyases required for growth on even and odd chain fatty acids. Here, we report that Mtb’s ICLs are essential for survival on both Results acetate and propionate because of its methylisocitrate lyase (MCL) Bactericidal Activity of Acetate and Propionate Against ICL-Deficient activity. Lack of MCL activity converts Mtb’s methylcitrate cycle Mtb. Previous studies reported that, consistent with predicted into a “dead end” pathway that sequesters tricarboxylic acid defects in the glyoxylate shunt and methylcitrate cycles, ICL- (TCA) cycle intermediates into methylcitrate cycle intermediates, deficient Mtb were specifically unable to grow when cultured in depletes gluconeogenic precursors, and results in defects of mem- media containing the model fatty acid metabolites, acetate or brane potential and intrabacterial pH. Activation of an alternative propionate (4–6).