High-Altitude Adaptation in Rhesus Macaques 4 5 Zachary A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Regulation of Xenobiotic and Bile Acid Metabolism by the Anti-Aging Intervention Calorie Restriction in Mice
REGULATION OF XENOBIOTIC AND BILE ACID METABOLISM BY THE ANTI-AGING INTERVENTION CALORIE RESTRICTION IN MICE By Zidong Fu Submitted to the Graduate Degree Program in Pharmacology, Toxicology, and Therapeutics and the Graduate Faculty of the University of Kansas in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Dissertation Committee ________________________________ Chairperson: Curtis Klaassen, Ph.D. ________________________________ Udayan Apte, Ph.D. ________________________________ Wen-Xing Ding, Ph.D. ________________________________ Thomas Pazdernik, Ph.D. ________________________________ Hao Zhu, Ph.D. Date Defended: 04-11-2013 The Dissertation Committee for Zidong Fu certifies that this is the approved version of the following dissertation: REGULATION OF XENOBIOTIC AND BILE ACID METABOLISM BY THE ANTI-AGING INTERVENTION CALORIE RESTRICTION IN MICE ________________________________ Chairperson: Curtis Klaassen, Ph.D. Date approved: 04-11-2013 ii ABSTRACT Calorie restriction (CR), defined as reduced calorie intake without causing malnutrition, is the best-known intervention to increase life span and slow aging-related diseases in various species. However, current knowledge on the exact mechanisms of aging and how CR exerts its anti-aging effects is still inadequate. The detoxification theory of aging proposes that the up-regulation of xenobiotic processing genes (XPGs) involved in phase-I and phase-II xenobiotic metabolism as well as transport, which renders a wide spectrum of detoxification, is a longevity mechanism. Interestingly, bile acids (BAs), the metabolites of cholesterol, have recently been connected with longevity. Thus, this dissertation aimed to determine the regulation of xenobiotic and BA metabolism by the well-known anti-aging intervention CR. First, the mRNA expression of XPGs in liver during aging was investigated. -

Flavin-Containing Monooxygenases: Mutations, Disease and Drug Response Phillips, IR; Shephard, EA
Flavin-containing monooxygenases: mutations, disease and drug response Phillips, IR; Shephard, EA For additional information about this publication click this link. http://qmro.qmul.ac.uk/jspui/handle/123456789/1015 Information about this research object was correct at the time of download; we occasionally make corrections to records, please therefore check the published record when citing. For more information contact [email protected] Flavin-containing monooxygenases: mutations, disease and drug response Ian R. Phillips1 and Elizabeth A. Shephard2 1School of Biological and Chemical Sciences, Queen Mary, University of London, Mile End Road, London E1 4NS, UK 2Department of Biochemistry and Molecular Biology, University College London, Gower Street, London WC1E 6BT, UK Corresponding author: Shephard, E.A. ([email protected]). and, thus, contribute to drug development. This review Flavin-containing monooxygenases (FMOs) metabolize considers the role of FMOs and their genetic variants in numerous foreign chemicals, including drugs, pesticides disease and drug response. and dietary components and, thus, mediate interactions between humans and their chemical environment. We Mechanism and structure describe the mechanism of action of FMOs and insights For catalysis FMOs require flavin adenine dinucleotide gained from the structure of yeast FMO. We then (FAD) as a prosthetic group, NADPH as a cofactor and concentrate on the three FMOs (FMOs 1, 2 and 3) that are molecular oxygen as a cosubstrate [5,6]. In contrast to most important for metabolism of foreign chemicals in CYPs FMOs accept reducing equivalents directly from humans, focusing on the role of the FMOs and their genetic NADPH and, thus, do not require accessory proteins. -

Hanna Joleen 2018 Thesis.Pdf
Validation of an In Vitro Mutagenicity Assay Based on Pulmonary Epithelial Cells from the Transgenic MutaMouse: Intra-Laboratory Variability and Metabolic Competence By: Joleen Hanna, B.Sc. A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science In Biology Specializing in Chemical and Environmental Toxicology Supervisor: Dr. Paul White (University of Ottawa) Thesis Advisory Committee: Dr. Frances Pick (University of Ottawa) Dr. Iain Lambert (Carleton University) University of Ottawa Ontario, Canada March 2018 © Joleen Hanna, Ottawa, Canada, 2018 Abstract: Genetic toxicity tests used for regulatory screening must be rigorously validated to ensure accuracy, reliability and relevance. Hence, prior to establishment of an internationally- accepted test guideline, a new assay must undergo multi-stage validation. An in vitro transgene mutagenicity assay based on an immortalized cell line derived from MutaMouse lung (i.e., FE1 cells) is currently undergoing formal validation. FE1 cells retain a lacZ transgene in a λgt10 shuttle vector that can be retrieved for scoring of chemically-induced mutations. This work contributes to validation of the in vitro transgene (lacZ) mutagenicity assay in MutaMouse FE1 cells. More specifically, the work includes an intra-laboratory variability study, and a follow-up study to assess the endogenous metabolic capacity of FE1 cells. The former is essential to determine assay reliability, the latter to define the range of chemicals that can be reliably screened without an exogenous metabolic activation mixture (i.e., rat liver S9). The intra- laboratory variability assessment revealed minimal variability; thus, assay reproducibility can be deemed acceptable. Assessment of metabolic capacity involved exposure of FE1 cells to 5 known mutagens, and subsequent assessment of changes in the expression of genes involved in xenobiotic metabolism; induced transgene mutant frequency (±S9) was assessed in parallel. -

Glucocorticoids Are Not Always Deleterious for Bone
CLINICAL VIGNETTE JBMR Glucocorticoids Are Not Always Deleterious for Bone Antoon HJM van Lierop, Neveen AT Hamdy, and Socrates E Papapoulos Department of Endocrinology and Metabolic Diseases, Leiden University Medical Center, Leiden, The Netherlands ABSTRACT A 23-year-old man with the rare sclerosing bone disorder van Buchem disease presented with progressively worsening headaches that eventually became persistent and associated with papilledema. Increased intracranial pressure was diagnosed, and the patient had a ventriculoperitoneal drain inserted as well as simultaneously receiving treatment with prednisone. Before starting treatment, there was biochemical evidence for increased bone turnover and for steady increases in bone mineral density (BMD) at the spine and total hip despite the patient having reached his peak height of 197 cm at the age of 19 years. Treatment with prednisone for 2 years resulted in biochemical and histologic suppression of bone formation as well as of bone resorption and arrest of further bone accumulation. Our data suggest that glucocorticoids (GCs) may represent an attractive alternative to the high-risk surgical approaches used in the management of patients with progressive sclerosing bone disorders. Our findings also suggest that whereas sclerostin may not be required for the action of GCs on bone formation, it may well be important for the action of GCs on bone resorption. The exact mechanism by which sclerostin may be involved in the regulation of bone resorption is as yet to be explored. ß 2010 American Society for Bone and Mineral Research. KEY WORDS: BONE RESORPTION; BONE FORMATION; VAN BUCHEM DISEASE; PREDNISONE; SCLEROSTIN Introduction We present here sequential observations of a patient with van Buchem disease with life-threatening increased intracranial an Buchem disease is a rare bone sclerosing disorder pressure who was treated successfully with prednisone. -

FMO3) As a Novel Genetic Determinant of Acetaminophen (APAP) Induced Hepatotoxicity Swetha Rudraiah University of Connecticut - Storrs, [email protected]
University of Connecticut OpenCommons@UConn Doctoral Dissertations University of Connecticut Graduate School 8-4-2014 Characterization of Flavin-containing Monooxygenase-3 (FMO3) as a Novel Genetic Determinant of Acetaminophen (APAP) Induced Hepatotoxicity Swetha Rudraiah University of Connecticut - Storrs, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/dissertations Recommended Citation Rudraiah, Swetha, "Characterization of Flavin-containing Monooxygenase-3 (FMO3) as a Novel Genetic Determinant of Acetaminophen (APAP) Induced Hepatotoxicity" (2014). Doctoral Dissertations. 506. https://opencommons.uconn.edu/dissertations/506 Characterization of Flavin-containing Monooxygenase-3 (FMO3) as a Novel Genetic Determinant of Acetaminophen (APAP) Induced Hepatotoxicity Swetha Rudraiah, Ph.D. University of Connecticut, 2014 Mice pretreated with a mild toxic dose of acetaminophen (APAP) acquire resistance to a second, higher APAP dose. This phenomenon is termed APAP autoprotection and the exact mechanism by which such resistance develops is not clearly known. Given the prevalence of APAP- hepatotoxicity and the human health impact of this potentially hepatotoxic agent, a further understanding of the mechanism(s) involved in such protection are of considerable significance and could lead to new modalities of treatment of acute drug-induced liver injury. The work presented in this thesis investigates FMO3 gene expression during APAP-induced liver injury as well as the functional significance of FMO3 over-expression during APAP- induced liver injury. Furthermore, FMO3 gene regulation during oxidative stress conditions is also examined. Acetaminophen treatment resulted in up-regulation of liver Fmo3 protein in male mice. Female mice express higher liver Fmo3 than males and are highly resistant to APAP hepatotoxicity. -

2018 Southern Regional Meeting
Abstracts J Investig Med: first published as 10.1136/jim-2017-000697.1 on 1 February 2018. Downloaded from Cardiovascular club I Purpose of study Coronary artery disease (C.A.D.) is one of the highest causes of death in the world. The purpose of this 11:00 AM study is to compare Puerto Rico (P.R.), Hispanic, U.S.A. coun- try, with the U.S.A., in coronary artery disease. Thursday, February 22, 2018 Methods used Compare a population of Hispanics with the high LDL levels with normal total cholesterol and HDL in P. R. and the U.S.A. The study population was 1000 patients. 1 A POSSIBLE ROLE FOR GENETICS IN CARDIOVASCULAR The U.S.A. health statistics and P.R. Department of Health DISEASE AMONG THE ACADIANS was used for comparison. AE Tedesco*, Z Haq, KL Di Losa, MJ Ali, P Gregory. LSUHSC – New Orleans, New Orleans, Summary of results Studying the lipid profile of Puerto Rico LA population, we found that the mean value of LDL lipoprotein is high (±104 mg/dl) with similar cholesterol and HDL levels 10.1136/jim-2017-000697.1 in both societies; still the coronary disease (CAD) incidence is lower than the U.S.A. (20%–30%). Investigators from the U.P. Purpose of study It is well documented that the Louisiana Aca- R. reported the genetic admixture of this Hispanic population. ‘ ’ dians ( Cajuns ) experience a disproportionate risk for some They reported the admixture consisted of 3 genes called pro- genetic diseases due to a genetic founder effect.Furthermore, cer- tective against C.A.D. -
Drosophila and Human Transcriptomic Data Mining Provides Evidence for Therapeutic
Drosophila and human transcriptomic data mining provides evidence for therapeutic mechanism of pentylenetetrazole in Down syndrome Author Abhay Sharma Institute of Genomics and Integrative Biology Council of Scientific and Industrial Research Delhi University Campus, Mall Road Delhi 110007, India Tel: +91-11-27666156, Fax: +91-11-27662407 Email: [email protected] Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 Running head: Pentylenetetrazole mechanism in Down syndrome 1 Abstract Pentylenetetrazole (PTZ) has recently been found to ameliorate cognitive impairment in rodent models of Down syndrome (DS). The mechanism underlying PTZ’s therapeutic effect is however not clear. Microarray profiling has previously reported differential expression of genes in DS. No mammalian transcriptomic data on PTZ treatment however exists. Nevertheless, a Drosophila model inspired by rodent models of PTZ induced kindling plasticity has recently been described. Microarray profiling has shown PTZ’s downregulatory effect on gene expression in fly heads. In a comparative transcriptomics approach, I have analyzed the available microarray data in order to identify potential mechanism of PTZ action in DS. I find that transcriptomic correlates of chronic PTZ in Drosophila and DS counteract each other. A significant enrichment is observed between PTZ downregulated and DS upregulated genes, and a significant depletion between PTZ downregulated and DS dowwnregulated genes. Further, the common genes in PTZ Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 downregulated and DS upregulated sets show enrichment for MAP kinase pathway. My analysis suggests that downregulation of MAP kinase pathway may mediate therapeutic effect of PTZ in DS. Existing evidence implicating MAP kinase pathway in DS supports this observation. -

Identification of Nondiabetic Heart Failure‑Associated Genes by Bioinformatics Approaches in Patients with Dilated Ischemic Cardiomyopathy
2602 EXPERIMENTAL AND THERAPEUTIC MEDICINE 11: 2602-2608, 2016 Identification of nondiabetic heart failure‑associated genes by bioinformatics approaches in patients with dilated ischemic cardiomyopathy ANZHONG YU1, JINGYAO ZHANG2, HAIYAN LIU3, BING LIU1 and LINGDONG MENG1 1Department of Cardiology, Jinan No. 4 People's Hospital, Jinan, Shandong 250031; 2Department of Blood Purification, Jinan Infectious Disease Hospital, Jinan, Shandong 250021; 3Department of Internal Medicine, Jinan Minzu Hospital, Jinan, Shandong 250014, P.R. China Received January 26, 2015; Accepted March 3, 2016 DOI: 10.3892/etm.2016.3252 Abstract. Heart failure (HF) is a common pathological conclusion, the two DEGs, NPPA and SERPINE1, may be condition affecting 4% of the worldwide population. important in the pathogenesis of HF and may be used for the However, approaches for predicting or treating nondiabetic diagnosis and treatment of HF. HF (ND-HF) progression are insufficient. In the current study, the gene expression profile GSE26887 was analyzed, Introduction which contained samples from 5 healthy controls, 7 diabetes mellitus-HF patients and 12 ND-HF patients with dilated Heart failure (HF), also known as chronic HF, is one of the ischemic cardiomyopathy. The dataset of 5 healthy controls major causes of mortality affecting approximately 4% of the and 12 ND-HF patients was normalized with robust multichip world's population, while the prevalence of this condition is average analysis and the differentially expressed genes (DEGs) currently increasing (1,2). In addition to the widely known were screened by unequal variance t-test and multiple-testing risk factor of glucose abnormalities (observed in ~43% of HF correction. In addition, the protein-protein interaction patients) (3), HF can also result from certain other factors, (PPI) network of the upregulated and downregulated genes which is classified as nondiabetic HF (ND‑HF) and is based was constructed using the Search Tool for the Retrieval of on a complicated pathological mechanism (4,5). -

Genetic Determinants of Brain Structure
UNIVERSITY OF CALIFORNIA Los Angeles Genetic Determinants of Brain Structure A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Engineering by Derrek Paul Hibar 2013 ! ABSTRACT OF THE DISSERTATION Genetic Determinants of Brain Structure by Derrek Paul Hibar Doctor of Philosophy in Biomedical Engineering University of California, Los Angeles, 2013 Professor Paul M. Thompson, Chair Over the past decade, billions of dollars from public and private funding institutions have been invested in the fields of neuroimaging and human genetics. Recently, researchers have realized that quantitative measures from imaging methods are a useful substrate for testing how genes influence brain structure, behavior, and susceptibility to disease. However, properly merging the two well-developed fields requires complex new methods and statistical models. Many studies in imaging genetics are simplistic in that they generally focus on individual association tests of a small set of genetic variants (usually single nucleotide polymorphisms, SNPs) on a single summary measure of the brain. These studies are great at bridging the gap between the two fields, but they often fail to utilize advanced methods from either field. For example, in genetics we know that our genes interact with each other in complex pathways, only ! in very rare circumstances is a single mutation or variant responsible for observable differences in phenotype. Modeling genetic associations using simple linear regression to test the effect of a single SNP at a time on an imaging phenotype is a good first step, but methods that more accurately represent the underlying biology will test the joint effect of multiple genetic variants simultaneously. -
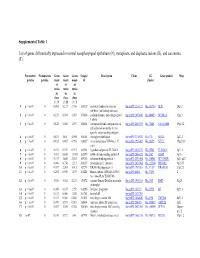
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

Consequences of Exchanging Carbohydrates for Proteins in the Cholesterol Metabolism of Mice Fed a High-Fat Diet
Consequences of Exchanging Carbohydrates for Proteins in the Cholesterol Metabolism of Mice Fed a High-fat Diet Fre´de´ ric Raymond1.¤a, Long Wang2., Mireille Moser1, Sylviane Metairon1¤a, Robert Mansourian1, Marie- Camille Zwahlen1, Martin Kussmann3,4,5, Andreas Fuerholz1, Katherine Mace´ 6, Chieh Jason Chou6*¤b 1 Bioanalytical Science Department, Nestle´ Research Center, Lausanne, Switzerland, 2 Department of Nutrition Science and Dietetics, Syracuse University, Syracuse, New York, United States of America, 3 Proteomics and Metabonomics Core, Nestle´ Institute of Health Sciences, Lausanne, Switzerland, 4 Faculty of Science, Aarhus University, Aarhus, Denmark, 5 Faculty of Life Sciences, Federal Institute of Technology, Lausanne, Switzerland, 6 Nutrition and Health Department, Nestle´ Research Center, Lausanne, Switzerland Abstract Consumption of low-carbohydrate, high-protein, high-fat diets lead to rapid weight loss but the cardioprotective effects of these diets have been questioned. We examined the impact of high-protein and high-fat diets on cholesterol metabolism by comparing the plasma cholesterol and the expression of cholesterol biosynthesis genes in the liver of mice fed a high-fat (HF) diet that has a high (H) or a low (L) protein-to-carbohydrate (P/C) ratio. H-P/C-HF feeding, compared with L-P/C-HF feeding, decreased plasma total cholesterol and increased HDL cholesterol concentrations at 4-wk. Interestingly, the expression of genes involved in hepatic steroid biosynthesis responded to an increased dietary P/C ratio by first down- regulation (2-d) followed by later up-regulation at 4-wk, and the temporal gene expression patterns were connected to the putative activity of SREBF1 and 2.