Physiology of Rumen Bacteria Associated with Low Methane Emitting Sheep
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Auxiliary Iron–Sulfur Cofactors in Radical SAM Enzymes☆
Biochimica et Biophysica Acta 1853 (2015) 1316–1334 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbamcr Review Auxiliary iron–sulfur cofactors in radical SAM enzymes☆ Nicholas D. Lanz a, Squire J. Booker a,b,⁎ a Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, United States b Department of Chemistry, The Pennsylvania State University, University Park, PA 16802, United States article info abstract Article history: A vast number of enzymes are now known to belong to a superfamily known as radical SAM, which all contain a Received 19 September 2014 [4Fe–4S] cluster ligated by three cysteine residues. The remaining, unligated, iron ion of the cluster binds in Received in revised form 15 December 2014 contact with the α-amino and α-carboxylate groups of S-adenosyl-L-methionine (SAM). This binding mode Accepted 6 January 2015 facilitates inner-sphere electron transfer from the reduced form of the cluster into the sulfur atom of SAM, Available online 15 January 2015 resulting in a reductive cleavage of SAM to methionine and a 5′-deoxyadenosyl radical. The 5′-deoxyadenosyl Keywords: radical then abstracts a target substrate hydrogen atom, initiating a wide variety of radical-based transforma- – Radical SAM tions. A subset of radical SAM enzymes contains one or more additional iron sulfur clusters that are required Iron–sulfur cluster for the reactions they catalyze. However, outside of a subset of sulfur insertion reactions, very little is known S-adenosylmethionine about the roles of these additional clusters. This review will highlight the most recent advances in the identifica- Cofactor maturation tion and characterization of radical SAM enzymes that harbor auxiliary iron–sulfur clusters. -

Bovine Host Genome Acts on Speci C Metabolism, Communication and Genetic Processes of Rumen Microbes Host-Genomically Linked To
Bovine host genome acts on specic metabolism, communication and genetic processes of rumen microbes host-genomically linked to methane emissions Marina Martínez-Álvaro SRUC https://orcid.org/0000-0003-2295-5839 Marc Auffret SRUC Carol-Anne Duthie SRUC Richard Dewhurst SRUC Matthew Cleveland Genus plc Mick Watson Roslin Institute https://orcid.org/0000-0003-4211-0358 Rainer Roehe ( [email protected] ) SRUC https://orcid.org/0000-0002-4880-3756 Article Keywords: bovine host genome, rumen, CH4 Posted Date: May 17th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-290150/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License 1 Bovine host genome acts on specific metabolism, communication and 2 genetic processes of rumen microbes host-genomically linked to methane 3 emissions 4 Marina Martínez-Álvaro1, Marc D. Auffret1, Carol-Anne Duthie1, Richard J. Dewhurst1, 5 Matthew A. Cleveland2, Mick Watson3 and Rainer Roehe*1 6 1Scotland’s Rural College, Edinburgh, UK 7 2Genus plc, DeForest, WI, USA 8 3The Roslin Institute and the Royal (Dick) School of Veterinary Studies, University of 9 Edinburgh, UK 10 11 *Corresponding author. Email: [email protected] 12 13 14 Introductory paragraph 15 Whereas recent studies in different species showed that the host genome shapes the microbial 16 community profile, our new research strategy revealed substantial host genomic control of 17 comprehensive functional microbial processes in the rumen of bovines by utilising microbial 18 gene profiles from whole metagenomic sequencing. Of 1,107/225/1,141 rumen microbial 19 genera/metagenome assembled uncultured genomes (RUGs)/genes identified, 203/16/352 20 were significantly (P<2.02 x10-5) heritable (0.13 to 0.61), revealing substantial variation in 21 host genomic control. -

Perilla Frutescens Leaf Alters the Rumen Microbial Community of Lactating Dairy Cows
microorganisms Article Perilla frutescens Leaf Alters the Rumen Microbial Community of Lactating Dairy Cows Zhiqiang Sun, Zhu Yu and Bing Wang * College of Grass Science and Technology, China Agricultural University, Beijing 100193, China; [email protected] (Z.S.); [email protected] (Z.Y.) * Correspondence: [email protected] Received: 25 September 2019; Accepted: 12 November 2019; Published: 13 November 2019 Abstract: Perilla frutescens (L.) Britt., an annual herbaceous plant, has antibacterial, anti-inflammation, and antioxidant properties. To understand the effects of P. frutescens leaf on the ruminal microbial ecology of cattle, Illumina MiSeq 16S rRNA sequencing technology was used. Fourteen cows were used in a randomized complete block design trial. Two diets were fed to these cattle: a control diet (CON); and CON supplemented with 300 g/d P. frutescens leaf (PFL) per cow. Ruminal fluid was sampled at the end of the experiment for microbial DNA extraction. Overall, our findings revealed that supplementation with PFL could increase ruminal fluid pH value. The ruminal bacterial community of cattle was dominated by Bacteroidetes, Firmicutes, and Proteobacteria. The addition of PFL had a positive effect on Firmicutes, Actinobacteria, and Spirochaetes, but had no effect on Bacteroidetes and Proteobacteria compared with the CON. The supplementation with PFL significantly increased the abundance of Marvinbryantia, Acetitomaculum, Ruminococcus gauvreauii, Eubacterium coprostanoligenes, Selenomonas_1, Pseudoscardovia, norank_f__Muribaculaceae, and Sharpea, and decreased the abundance of Treponema_2 compared to CON. Eubacterium coprostanoligenes, and norank_f__Muribaculaceae were positively correlated with ruminal pH value. It was found that norank_f__Muribaculaceae and Acetitomaculum were positively correlated with milk yield, indicating that these different genera are PFL associated bacteria. -

( 12 ) United States Patent
US010435714B2 (12 ) United States Patent ( 10 ) Patent No. : US 10 ,435 ,714 B2 Gill et al. (45 ) Date of Patent : * Oct. 8 , 2019 (54 ) NUCLEIC ACID -GUIDED NUCLEASES 8 ,569 ,041 32 10 / 2013 Church et al. 8 ,697 , 359 B1 4 / 2014 Zhang 8 , 906 ,616 B2 12 / 2014 Zhang et al. ( 71 ) Applicant: Inscripta , Inc. , Boulder, CO (US ) 9 , 458 ,439 B2 10 / 2016 Choulika et al . 9 ,512 ,446 B112 /2016 Joung et al. (72 ) Inventors : Ryan T . Gill, Denver , CO (US ) ; 9 ,752 , 132 B2 9 / 2017 Joung et al. Andrew Garst , Boulder , CO (US ) ; 9 , 790 , 490 B2 10 / 2017 Zhang et al. Tanya Elizabeth Warnecke Lipscomb, 9 ,926 ,546 B2 3 / 2018 Joung et al . 9 , 982 ,278 B2 5 /2018 Gill et al. Boulder, CO (US ) 9 , 982 ,279 B1 5 / 2018 Gill et al. 10 ,011 , 849 B1 7 / 2018 Gill et al . ( 73 ) Assignee : INSCRIPTA , INC ., Boulder, CO (US ) 10 ,017 , 760 B2 7 / 2018 Gill et al . 2008 /0287317 AL 11/ 2008 Boone ( * ) Notice : Subject to any disclaimer , the term of this 2009 /0176653 Al 7 / 2009 Kim et al. patent is extended or adjusted under 35 2010 /0034924 Al 2 / 2010 Fremaux et al . 2010 / 0305001 A1 12 /2010 Kern et al . U .S . C . 154 (b ) by 48 days . 2014 / 0068797 A1 3 / 2014 Doudna et al . 2014 / 0089681 A1 3 / 2014 Goto et al. This patent is subject to a terminal dis 2014 /0121118 A1 5 / 2014 Warner claimer . 2014 / 0199767 A1 7 /2014 Barrangou et al. -

A Microbiologist's View on Improving Nutrient Utilization in Ruminants
A Microbiologist’s View on Improving Nutrient Utilization in Ruminants T. G. Nagaraja1 College of Veterinary Medicine, Kansas State University, Introduction Ruminants, particularly cattle, sheep, and goats, are important production animals for food to humans worldwide. Their importance comes from their unique ability to convert, because of foregut microbial fermentation, fiber-based feeds with or without grains, into high quality, protein-rich products like milk and meat. The rumen, the first compartment of the complex stomach, is inhabited by a multitude of microbes that work in concert to breakdown feeds to produce energy (volatile fatty acids; VFA), protein (microbial cells) and other nutrients like vitamins (microbial cells) to the host. The production of VFA, mainly from carbohydrates. is central to the ruminal fermentation because the process provides energy (ATP) for microbial growth, which serves as the major source of protein to the host, but also provides the animal with the precursors necessary to generate energy (mainly acetate), glucose (mainly propionate), and lipid (mainly acetate and butyrate). The fermentation of nitrogenous compounds is also an integral process because it provides the molecules necessary to build microbial cell protein. In addition to the importance of the rumen microbial function to the host nutrition and food production, rumen microbes and their enzymes are also of considerable interest to the biofuels and biotechnology industries (Hess et al., 2011). Despite the tremendous importance, rumen remains an under investigated, hence, under-characterized, microbial ecosystem. At one time, rumen was the most extensively investigated anaerobic ecosystem. In the past 10 to 12 years, human gut microbial studies have far outpaced rumen microbiology studies. -

Effects of Long-Acting, Broad Spectra Anthelmintic Treatments on The
www.nature.com/scientificreports OPEN Efects of long‑acting, broad spectra anthelmintic treatments on the rumen microbial community compositions of grazing sheep Christina D. Moon1*, Luis Carvalho1, Michelle R. Kirk1, Alan F. McCulloch2, Sandra Kittelmann3, Wayne Young1, Peter H. Janssen1 & Dave M. Leathwick1 Anthelmintic treatment of adult ewes is widely practiced to remove parasite burdens in the expectation of increased ruminant productivity. However, the broad activity spectra of many anthelmintic compounds raises the possibility of impacts on the rumen microbiota. To investigate this, 300 grazing ewes were allocated to treatment groups that included a 100‑day controlled release capsule (CRC) containing albendazole and abamectin, a long‑acting moxidectin injection (LAI), and a non‑treated control group (CON). Rumen bacterial, archaeal and protozoal communities at day 0 were analysed to identify 36 sheep per treatment with similar starting compositions. Microbiota profles, including those for the rumen fungi, were then generated for the selected sheep at days 0, 35 and 77. The CRC treatment signifcantly impacted the archaeal community, and was associated with increased relative abundances of Methanobrevibacter ruminantium, Methanosphaera sp. ISO3‑F5, and Methanomassiliicoccaceae Group 12 sp. ISO4‑H5 compared to the control group. In contrast, the LAI treatment increased the relative abundances of members of the Veillonellaceae and resulted in minor changes to the bacterial and fungal communities by day 77. Overall, the anthelmintic treatments resulted in few, but highly signifcant, changes to the rumen microbiota composition. Anthelmintic treatment of adult ewes is practiced by about 80% of sheep farmers in New Zealand1, and many of these treatments use active compounds with broad-spectrum persistent activity. -

Changes in Soil Microbial Communities After Long-Term Warming Exposure" (2019)
University of Massachusetts Amherst ScholarWorks@UMass Amherst Doctoral Dissertations Dissertations and Theses October 2019 CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG- TERM WARMING EXPOSURE William G. Rodríguez-Reillo University of Massachusetts Amherst Follow this and additional works at: https://scholarworks.umass.edu/dissertations_2 Part of the Environmental Microbiology and Microbial Ecology Commons Recommended Citation Rodríguez-Reillo, William G., "CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE" (2019). Doctoral Dissertations. 1757. https://doi.org/10.7275/15007293 https://scholarworks.umass.edu/dissertations_2/1757 This Open Access Dissertation is brought to you for free and open access by the Dissertations and Theses at ScholarWorks@UMass Amherst. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected]. CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE A Dissertation Presented by WILLIAM GABRIEL RODRÍGUEZ-REILLO Submitted to the Graduate School of the University of Massachusetts Amherst in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY SEPTEMBER 2019 Organismic and Evolutionary Biology © Copyright by William Gabriel Rodríguez-Reillo 2019 All Rights Reserved CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE A Dissertation Presented by WILLIAM GABRIEL RODRÍGUEZ-REILLO Approved as to style and content by: _________________________________________ Jeffrey L. Blanchard, Chair _________________________________________ Courtney Babbitt, Member _________________________________________ David Sela, Member _________________________________________ Kristina Stinson, Member ______________________________________ Paige Warren, Graduate Program Director Organismic and Evolutionary Biology DEDICATION To my parents, William Rodriguez Arce and Carmen L. Reillo Batista. A quienes aún en la distancia me mantuvieron en sus oraciones. -

Effects of Starter Feeds of Different Physical Forms on Rumen Fermentation and Microbial Composition for Pre-Weaning and Post-We
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.03.235580; this version posted August 4, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Effects of Starter Feeds of Different Physical Forms on Rumen Fermentation and 2 Microbial Composition for Pre-weaning and Post-weaning Lambs 3 4 Yong Li,* Yanli Guo,# Chengxin Zhang, Xiaofang Cai, Peng Liu, Cailian Li 5 6 College of Animal Science and Technology, Gansu Agricultural University, Lanzhou 7 730070, P.R. China 8 9 10 11 12 13 14 15 16 17 18 19 #Address correspondence to Yanli Guo, [email protected]. 20 *Present address: Yong Li, Zhoukou Vocational and Technical College, Zhoukou, P. R. 21 China. 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.08.03.235580; this version posted August 4, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 22 ABSTRACT 23 This study aimed to evaluate the effects of starter feeds of different physical forms 24 on rumen fermentation and microbial composition for lambs. Twenty-four eight-day-old 25 male Hu lambs (5.04 ± 0.75 kg body weight) were fed either milk replacer (MR) and 26 pelleted starter feed (PS), or MR and textured starter feed (TS) in pre-weaning (day 8 to 27 35) and post-weaning (day 36 to 42) lambs. -

Metabolic Functions of the Human Gut Microbiota: the Role of Metalloenzymes Cite This: DOI: 10.1039/C8np00074c Lauren J
Natural Product Reports View Article Online REVIEW View Journal Metabolic functions of the human gut microbiota: the role of metalloenzymes Cite this: DOI: 10.1039/c8np00074c Lauren J. Rajakovich and Emily P. Balskus * Covering: up to the end of 2017 The human body is composed of an equal number of human and microbial cells. While the microbial community inhabiting the human gastrointestinal tract plays an essential role in host health, these organisms have also been connected to various diseases. Yet, the gut microbial functions that modulate host biology are not well established. In this review, we describe metabolic functions of the human gut microbiota that involve metalloenzymes. These activities enable gut microbial colonization, mediate interactions with the host, and impact human health and disease. We highlight cases in which enzyme characterization has advanced our understanding of the gut microbiota and examples that illustrate the diverse ways in which Received 14th August 2018 Creative Commons Attribution 3.0 Unported Licence. metalloenzymes facilitate both essential and unique functions of this community. Finally, we analyze Human DOI: 10.1039/c8np00074c Microbiome Project sequencing datasets to assess the distribution of a prominent family of metalloenzymes rsc.li/npr in human-associated microbial communities, guiding future enzyme characterization efforts. 1 Introduction our gut from infancy and plays a critical role in the development 2 Commensal colonization of the human gut: tness and and maintenance of healthy human physiology. It aids in adaptation developing the innate and adaptive immune systems,1,2 2.1 Glycan sulfation provides nutrients and vitamins,3 and protects against path- 4 This article is licensed under a 2.2 Glycan fucosylation ogen invasion. -

2018 Summer Symposium Program
Ofce of UNDERGRADUATE RESEARCH ® THE UNIVERSITY OF UTAH 2018 Summer Symposium THURSDAY, AUGUST 2, 2018 9:00AM - 12:00PM CROCKER SCIENCE CENTER UNIVERSITY OF UTAH 2018 SUMMER SYMPOSIUM Thursday, August 2, 2018 9:00 AM – 12:00 PM Crocker Science Center University of Utah The Office of Undergraduate Research is grateful for the generous support of the Office of the Vice President for Research. We are also thankful for the development of the Summer Research Program Partnership, which is a new collaboration among the Chemistry Research Experience for Undergraduates (REU), the Huntsman Cancer Institute’s PathMaker Cancer Research Program, the Native American Summer Research Internship (NARI), the Physics & Astronomy REU and Summer Undergraduate Research Program, and the Summer Program for Undergraduate Research (SPUR). Together, these programs are serving more than 90 undergraduate researchers in Summer 2018. Finally, we would like to express our utmost pride and congratulations to the students, graduate students, and faculty mentors without whose efforts and dedication this event would not be possible. PROGRAM SCHEDULE NOTE: All student presenters MUST check-in Snacks available at 10:15 AM in Room 205/206 8:30 – 9:00 AM CHECK-IN & POSTER SET-UP 9:00 – 10:30 AM POSTER SESSION I (ODD POSTERS) 10:30 AM – 12:00 PM POSTER SESSION II (EVEN POSTERS) 12:00 – 12:15 PM POSTER TAKE-DOWN 2 SCHEDULE OF PRESENTATIONS POSTER SESSION I 9:00 – 10:30 AM Poster 1 Presenter: Sara Alektiar (University of Michigan) Mentor: Matthew Sigman (Chemistry) Electrocatalytic Bis(bipyidine)ruthenium Hydroxylation of Tertiary and Benzylic C-H Bonds The Sigman and Du Bois labs recently reported a methodology that employs a bis(bipyridine)Ru catalyst operating in acidic water to achieve oxidation of tertiary and benzylic C-H bonds in the presence of basic amines. -
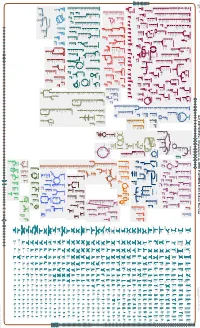
Generated by SRI International Pathway Tools Version 25.0 on Mon
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000716605Cyc: Streptomyces sp. NRRL F-525 NRRL F-525 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari an amino an amino an amino an amino an amino an amino an amino an amino phosphate phosphate acid acid acid acid acid acid acid acid molybdate an amino an amino H + a dipeptide phosphate phosphate phosphate acid an amino acid an amino H + phosphate phosphate glycine betaine glycine betaine glycine betaine leu pro pro pro acid acid predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted ABC ABC ABC ABC ABC ABC ABC ABC ABC ABC ABC F0F1 ATP ABC RS16420 RS10685 RS06340 RS46495 RS16425 ProP RS28920 RS26410 LeuE RS27350 RS38790 RS13800 transporter transporter transporter RS41005 transporter transporter transporter transporter transporter CoxB transporter transporter transporter synthase transporter of an of an of an of an of an of an of an of an of molybdate of phosphate of phosphate of a dipeptide amino acid -

Notice: Restrictions
NOTICE: The copyright law of the United States (Title 17, United States Code) governs the making of reproductions of copyrighted material. One specified condition is that the reproduction is not to be "used for any purpose other than private study, scholarship, or research." If a user makes a request for, or later uses a reproduction for purposes in excess of "fair use," that user may be liable for copyright infringement. RESTRICTIONS: This student work may be read, quoted from, cited, for purposes of research. It may not be published in full except by permission of the author. Abstract: The phylogenetic position of a number of bacteria within the family Flavobacteriaceae has been questioned. To address the question, the whole genomes of several organisms were sequenced, and this project is focused on Chryseobacterium haifense. The advances in next generation sequencing (NGS) technologies have caused a decrease in cost for whole genome sequencing. This decreased cost has led to more genomes being sequenced and in the process has caused a large demand for bioinformatics tools to handle the genomic data. To analyze the genomic data, the 930,000 reads were assembled in several steps, using several different software packages to refine the assembly to fewer than 700 contiguous sequences. Automated annotation using the Rapid Annotation using Subsystem Technologies (RAST) server identified the organism’s genes and known pathways which were compared to its phenotypes. The Reciprocal Orthology Score Average (ROSA) genomic similarity calculator showed that Chryseobacterium haifense is as different from “true” Chryseobacteria as other separate genera are which has led to the conclusion that Chryseobacterium haifense does not belong within the Chryseobacterium genus.