Incipient Genome Erosion and Metabolic Streamlining for Antibiotic Production in a Defensive Symbiont
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

METACYC ID Description A0AR23 GO:0004842 (Ubiquitin-Protein Ligase
Electronic Supplementary Material (ESI) for Integrative Biology This journal is © The Royal Society of Chemistry 2012 Heat Stress Responsive Zostera marina Genes, Southern Population (α=0. -

Genome Mining of Streptomyces Formicae KY5 for Potential Drug Like Natural Products Characterizations
ics OPEN ACCESS Freely available online om & B te i ro o P in f f o o r l m a a Journal of n t r i c u s o J ISSN: 0974-276X Proteomics & Bioinformatics Research Article Genome Mining of Streptomyces formicae KY5 for Potential Drug like Natural Products Characterizations Mohibullah Shah1*, Sana Gul2, Adnan Amjad3, Muhammad Sameem Javed3, Batool Fatima1, Haq Nawaz1, Jaweria Ishaq1 1Department of Biochemistry, Bahauddin Zakariya University, Multan-66000, Pakistan; 2Department of Biochemistry, Abdul Wali Khan University Mardan, Mardan-23000, Pakistan; 3Institute of Food Science and Nutrition, Bahauddin Zakariya University, Multan-66000, Pakistan ABSTRACT Genus Streptomyces has been a source of various clinically significant bioactive metabolites. Taxonomically, Streptomyces formicae KY5 is a new and different species. The complete genome sequences of S. formicae KY5 is available in the public DNA sequence databases for different analysis. The accessibility of the genomic sequence presents an excellent opportunity to explore the secondary metabolites potential of this distinct Streptomyces species. In this study, we employed the advance bioinformatics resources to annotate the total genome sequence of S. formicae KY5. Bioinformatics tools are applied to locate all the secondary metabolites hiding beneath their biosynthetic gene clusters (BGCs). The S. formicae KY5 is found to synthesis distinct and various secondary metabolites by undergoing the designated genomic encoding. Predictive analysis conveys that this strain has 34 gene clusters to encode potential secondary metabolites. For structural similarity with other drugs, we scanned the drug bank database, drug target and drug with the highest similarity was retrieved from PDB for molecular docking. -

The Transcriptional Landscape of a Rewritten Bacterial Genome Reveals Control Elements and Genome Design Principles ✉ ✉ Mariëlle J
ARTICLE https://doi.org/10.1038/s41467-021-23362-y OPEN The transcriptional landscape of a rewritten bacterial genome reveals control elements and genome design principles ✉ ✉ Mariëlle J. F. M. van Kooten 1 , Clio A. Scheidegger1, Matthias Christen1 & Beat Christen 1 Sequence rewriting enables low-cost genome synthesis and the design of biological systems with orthogonal genetic codes. The error-free, robust rewriting of nucleotide sequences can 1234567890():,; be achieved with a complete annotation of gene regulatory elements. Here, we compare transcription in Caulobacter crescentus to transcription from plasmid-borne segments of the synthesized genome of C. ethensis 2.0. This rewritten derivative contains an extensive amount of supposedly neutral mutations, including 123’562 synonymous codon changes. The tran- scriptional landscape refines 60 promoter annotations, exposes 18 termination elements and links extensive transcription throughout the synthesized genome to the unintentional intro- duction of sigma factor binding motifs. We reveal translational regulation for 20 CDS and uncover an essential translational regulatory element for the expression of ribosomal protein RplS. The annotation of gene regulatory elements allowed us to formulate design principles that improve design schemes for synthesized DNA, en route to a bright future of iteration- free programming of biological systems. 1 Institute of Molecular Systems Biology, Department of Biology, Eidgenössische Technische Hochschule Zürich, Zürich, Switzerland. ✉ email: [email protected]; [email protected] NATURE COMMUNICATIONS | (2021) 12:3053 | https://doi.org/10.1038/s41467-021-23362-y | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-021-23362-y e can program biological systems with DNA that is In previous work, we synthesized and assembled the rewritten Wbased on native nucleotide sequences. -

The Pathogenomics and Evolution of Anthrax-Like Bacillus Cereus Isolates and Plasmids
The Pathogenomics and Evolution of Anthrax-like Bacillus cereus Isolates and Plasmids A white paper proposal submitted by: Geraldine A. Van der Auwera, Ph.D. Harvard Medical School Michael Feldgarden, Ph.D. Genomic Sequencing Center for Infectious Diseases The Broad Institute of MIT and Harvard 1 Executive summary A key member of the Bacillus cereus group, Bacillus anthracis is defined by phenotypic and molecular characteristics that are conferred by two large plasmids, pXO1 and pXO2. However the very concept of B. anthracis as a distinct species has been called into question by recent discoveries of “intermediate” isolates identified as B. cereus and B. thuringiensis but possessing features similar to those of B. anthracis, including large plasmids that share a common backbone with pXO1 and/or pXO2. Many of these “intermediate” isolates possess potential or demonstrated lethal pathogenic properties and are sometimes called “anthrax-like”, even though they do not meet the strict definition of anthrax-causing B. anthracis. We recently showed that pXO1- and pXO2- like plasmids are widely prevalent in environmental isolates of the B. cereus group. Because B. anthracis-like isolates do not possess all the molecular hallmarks of typical B. anthracis, there is a significant risk that they would escape being flagged as dangerous. Consequently, accidental infection by naturally occurring pathotypes which are not immediately recognized as life-threatening could present a serious health concern. Such cases have already been reported, some with a fatal outcome. The second risk posed by these B. anthracis-like isolates could be the intentional use as “stealth anthrax” bioweapon, either in natural form or with genetic modifications that would require only minimal skills and facilities to produce. -

Mapping Active Site Residues in Glutamate-5-Kinase. the Substrate Glutamate and the Feed-Back Inhibitor Proline Bind at Overlapping Sites
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 580 (2006) 6247–6253 Mapping active site residues in glutamate-5-kinase. The substrate glutamate and the feed-back inhibitor proline bind at overlapping sites Isabel Pe´rez-Arellanoa, Vicente Rubiob, Javier Cerveraa,* a Centro de Investigacio´n Prı´ncipe Felipe, Avda. Autopista del Saler, 16, Valencia 46013, Spain b Instituto de Biomedicina de Valencia (IBV-CSIC), Jaime Roig 11, Valencia 46010, Spain Received 22 August 2006; revised 10 October 2006; accepted 12 October 2006 Available online 20 October 2006 Edited by Stuart Ferguson (a domain named after pseudo uridine synthases and archaeo- Abstract Glutamate-5-kinase (G5K) catalyzes the controlling first step of proline biosynthesis. Substrate binding, catalysis sine-specific transglycosylases) domain [10]. AAK domains and feed-back inhibition by proline are functions of the N-termi- bind ATP and a phosphorylatable acidic substrate, and make nal 260-residue domain of G5K. We study here the impact on a phosphoric anhydride [11]. The function of the PUA domain these functions of 14 site-directed mutations affecting 9 residues (a domain characteristically found in some RNA-modifying of Escherichia coli G5K, chosen on the basis of the structure of enzymes) [12] is not known. By deleting the PUA domain of the bisubstrate complex of the homologous enzyme acetylgluta- E. coli G5K we showed [10] that the isolated AAK domain, mate kinase (NAGK). The results support the predicted roles as the complete enzyme, forms tetramers, catalyzes the reac- of K10, K217 and T169 in catalysis and ATP binding and of tion and is feed-back inhibited by proline. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -

The Functions and Evolution of Social Fluid Exchange in Ant Colonies (Hymenoptera: Formicidae) Marie-Pierre Meurville & Adria C
ISSN 1997-3500 Myrmecological News myrmecologicalnews.org Myrmecol. News 31: 1-30 doi: 10.25849/myrmecol.news_031:001 13 January 2021 Review Article Trophallaxis: the functions and evolution of social fluid exchange in ant colonies (Hymenoptera: Formicidae) Marie-Pierre Meurville & Adria C. LeBoeuf Abstract Trophallaxis is a complex social fluid exchange emblematic of social insects and of ants in particular. Trophallaxis behaviors are present in approximately half of all ant genera, distributed over 11 subfamilies. Across biological life, intra- and inter-species exchanged fluids tend to occur in only the most fitness-relevant behavioral contexts, typically transmitting endogenously produced molecules adapted to exert influence on the receiver’s physiology or behavior. Despite this, many aspects of trophallaxis remain poorly understood, such as the prevalence of the different forms of trophallaxis, the components transmitted, their roles in colony physiology and how these behaviors have evolved. With this review, we define the forms of trophallaxis observed in ants and bring together current knowledge on the mechanics of trophallaxis, the contents of the fluids transmitted, the contexts in which trophallaxis occurs and the roles these behaviors play in colony life. We identify six contexts where trophallaxis occurs: nourishment, short- and long-term decision making, immune defense, social maintenance, aggression, and inoculation and maintenance of the gut microbiota. Though many ideas have been put forth on the evolution of trophallaxis, our analyses support the idea that stomodeal trophallaxis has become a fixed aspect of colony life primarily in species that drink liquid food and, further, that the adoption of this behavior was key for some lineages in establishing ecological dominance. -
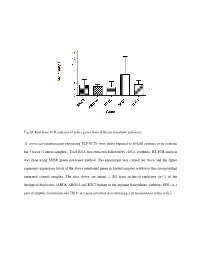
Fig.S1 Real-Time PCR Analysis of Select Genes from Different Metabolic Pathways S. Cerevisiae Transformants Expressing TEF-YCT1
Fig.S1 Real-time PCR analysis of select genes from different metabolic pathways S. cerevisiae transformants expressing TEF-YCT1 were either exposed to 500µM cysteine or no cysteine for 5 hours (Control samples). Total RNA was extracted followed by cDNA synthesis. RT-PCR analysis was done using SYBR green dye-based method. The experiment was carried out twice and the figure represents expression levels of the above mentioned genes in treated samples relative to the corresponding untreated control samples. The data above are means ± SD from technical replicates (n=3) of the biological duplicates. [ARG8, ARG5,6 and RTC2 belong to the arginine biosynthetic pathway, SSU1 is a part of sulphur metabolism and TIS11 is a gene involved in maintaining iron homeostasis in the cells.] Table S1: List of strains used in this study Strain Genotype Source ABC 1738 (yct1) MATα his31 leu20 lys20 ura30 YLL055W:: kanMX4 Euroscarf (Y01543) ABC3612(arg3) MATa his31 leu20 ura30 YJL088w::kanMX4 Euroscarf (Y11335) ABC4812(arg5,6) MATa his31 leu20 ura30 YER069w::kanMX4 Euroscarf (Y10209) ABC4811(arg8) MATa his31 leu20 ura30 YOL140w::kanMX4 Euroscarf (Y17711) ABC4814(spe1) MATa his31 leu20 ura30 YKL184w::kanMX4 Euroscarf (Y15034) ABC4815(spe2) MATa his31 leu20 ura30 YOL052c::kanMX4 Euroscarf (Y11743) ABC4816(spe3) MATa his31 leu20 ura30 YPR069c::kanMX4 Euroscarf (Y15488) ABC4817(spe4) MATa his31 leu20 ura30 YLR146c::kanMX4 Euroscarf (Y16945) ABC3604(ssu1) MATa his31 leu20 ura30 YPL092w::kanMX4 Euroscarf (Y12160) ABC1486(str2) MATa his31 leu20 ura30 YJR130c::kanMX4 -

Horizontal Gene Flow Into Geobacillus Is Constrained by the Chromosomal Organization of Growth and Sporulation
bioRxiv preprint doi: https://doi.org/10.1101/381442; this version posted August 2, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Horizontal gene flow into Geobacillus is constrained by the chromosomal organization of growth and sporulation Alexander Esin1,2, Tom Ellis3,4, Tobias Warnecke1,2* 1Molecular Systems Group, Medical Research Council London Institute of Medical Sciences, London, United Kingdom 2Institute of Clinical Sciences, Faculty of Medicine, Imperial College London, London, United Kingdom 3Imperial College Centre for Synthetic Biology, Imperial College London, London, United Kingdom 4Department of Bioengineering, Imperial College London, London, United Kingdom *corresponding author ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/381442; this version posted August 2, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Horizontal gene transfer (HGT) in bacteria occurs in the context of adaptive genome architecture. As a consequence, some chromosomal neighbourhoods are likely more permissive to HGT than others. Here, we investigate the chromosomal topology of horizontal gene flow into a clade of Bacillaceae that includes Geobacillus spp. Reconstructing HGT patterns using a phylogenetic approach coupled to model-based reconciliation, we discover three large contiguous chromosomal zones of HGT enrichment. -

14S802 - Strain Specific Pathogenicity of Staphylococcus Aureus Final Report
14S802 - Strain Specific Pathogenicity of Staphylococcus aureus Final Report This project was funded under the Department of Agriculture, Food and the Marine Competitive Funding Programme. SUMMARY Mastitis is a costly endemic disease for the dairy industry. It is primarily caused by bacterial infection and is the most common reason for antibiotic use in dairy cows in Ireland. Staphylococcus aureus is the most common mastitis pathogen in Ireland and the S. aureus strains that cause mastitis belong to specific bovine-adapted lineages. Current selection for mastitis-resistance is based on the host immune response, as determined by somatic cell count (SCC). However, the ability of S. aureus to evade and subvert the host immune response is well known, including the ability to internalise and survive within host cells. This project tested the hypothesis that bovine intramammary infection with different S. aureus strains results in differential activation of the host immune response. Supporting this hypothesis, significant differences between S. aureus lineages in their ability to internalise within bovine mammary epithelial cells were found with some strains internalising at higher levels than others. It was also found that some strains induced higher expression of cytokines and chemokines responsible for attracting immune cells and these strains induced mammary epithelial cells to produce factors that attracted somatic cells, while other strains did not. Differences in disease presentation in vivo in cows infected with different strains were also observed, indicating strain-specific virulence. Significantly higher somatic cell count and anti- Staphylococcus IgG and significantly lower milk yield were observed in response to infection with a more virulent strain. -

Table S5. the Information of the Bacteria Annotated in the Soil Community at Species Level
Table S5. The information of the bacteria annotated in the soil community at species level No. Phylum Class Order Family Genus Species The number of contigs Abundance(%) 1 Firmicutes Bacilli Bacillales Bacillaceae Bacillus Bacillus cereus 1749 5.145782459 2 Bacteroidetes Cytophagia Cytophagales Hymenobacteraceae Hymenobacter Hymenobacter sedentarius 1538 4.52499338 3 Gemmatimonadetes Gemmatimonadetes Gemmatimonadales Gemmatimonadaceae Gemmatirosa Gemmatirosa kalamazoonesis 1020 3.000970902 4 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas indica 797 2.344876284 5 Firmicutes Bacilli Lactobacillales Streptococcaceae Lactococcus Lactococcus piscium 542 1.594633558 6 Actinobacteria Thermoleophilia Solirubrobacterales Conexibacteraceae Conexibacter Conexibacter woesei 471 1.385742446 7 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas taxi 430 1.265115184 8 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas wittichii 388 1.141545794 9 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas sp. FARSPH 298 0.876754244 10 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sorangium cellulosum 260 0.764953367 11 Proteobacteria Deltaproteobacteria Myxococcales Polyangiaceae Sorangium Sphingomonas sp. Cra20 260 0.764953367 12 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas panacis 252 0.741416341 -

Section 4. Guidance Document on Horizontal Gene Transfer Between Bacteria
306 - PART 2. DOCUMENTS ON MICRO-ORGANISMS Section 4. Guidance document on horizontal gene transfer between bacteria 1. Introduction Horizontal gene transfer (HGT) 1 refers to the stable transfer of genetic material from one organism to another without reproduction. The significance of horizontal gene transfer was first recognised when evidence was found for ‘infectious heredity’ of multiple antibiotic resistance to pathogens (Watanabe, 1963). The assumed importance of HGT has changed several times (Doolittle et al., 2003) but there is general agreement now that HGT is a major, if not the dominant, force in bacterial evolution. Massive gene exchanges in completely sequenced genomes were discovered by deviant composition, anomalous phylogenetic distribution, great similarity of genes from distantly related species, and incongruent phylogenetic trees (Ochman et al., 2000; Koonin et al., 2001; Jain et al., 2002; Doolittle et al., 2003; Kurland et al., 2003; Philippe and Douady, 2003). There is also much evidence now for HGT by mobile genetic elements (MGEs) being an ongoing process that plays a primary role in the ecological adaptation of prokaryotes. Well documented is the example of the dissemination of antibiotic resistance genes by HGT that allowed bacterial populations to rapidly adapt to a strong selective pressure by agronomically and medically used antibiotics (Tschäpe, 1994; Witte, 1998; Mazel and Davies, 1999). MGEs shape bacterial genomes, promote intra-species variability and distribute genes between distantly related bacterial genera. Horizontal gene transfer (HGT) between bacteria is driven by three major processes: transformation (the uptake of free DNA), transduction (gene transfer mediated by bacteriophages) and conjugation (gene transfer by means of plasmids or conjugative and integrated elements).