The Laboratory in the Multidisciplinary Diagnosis of Differences Or
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Accepted Preprint First Posted on 4 July 2018 As Manuscript EJE-18
Page 1 of 26 Accepted Preprint first posted on 4 July 2018 as Manuscript EJE-18-0256 Title page Title: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): position paper of EU COST Action BM 1303 "DSDnet" Authors: L. Audí 1, S.F. Ahmed 2, N. Krone 3, M. Cools 4, K. McElreavey 5, P.M. Holterhus 6, A. Greenfield 7, A. Bashamboo 5, O. Hiort 8, S.A. Wudy 9, R. McGowan 2,10, on behalf of the EU COST Action 1 Growth and Development Research Unit, Vall d’Hebron Research Institute (VHIR), Center for Biomedical Research on Rare Diseases (CIBERER), Instituto de Salud Carlos III, Barcelona, 08035, Spain 2 Developmental Endocrinology Research Group, University of Glasgow, Glasgow, UK 3 Academic Unit of Child Health, Department of Oncology and Metabolism, University of Sheffield, Sheffield Children's Hospital, Western Bank, Sheffield S10 2TH, United Kingdom 4 Department of Paediatric Endocrinology, Ghent University Hospital, Paediatrics and Internal Medicine Research Unit, Ghent University, 9000 Ghent, Belgium 5 Human Developmental Genetics, Institut Pasteur, Paris, France 6 Division of Pediatric Endocrinology and Diabetes, University Hospital of Schleswig-Holstein and Christian Albrechts University, Kiel, Germany 7 Mammalian Genetics Unit, Medical Research Council, Harwell Institute, Oxfordshire, UK 1 Copyright © 2018 European Society of Endocrinology. Page 2 of 26 8 Division of Paediatric Endocrinology and Diabetes. Department of Paediatric and Adolescent Medicine, University of Lübeck, Lübeck, Germany 9 Steroid Research & Mass Spectrometry Unit, Laboratory for Translational Hormone Analytics, Division of Pediatric Endocrinology and Diabetology, Center of Child and Adolescent Medicine, Justus-Liebig-University, Giessen, Germany 10 Department of Clinical Genetics, Laboratories Building, Queen Elizabeth University Hospital, Glasgow, UK Corresponding author: Laura Audí Vall d’Hebron Research Institute (VHIR)- P. -

ACAT) in Cholesterol Metabolism: from Its Discovery to Clinical Trials and the Genomics Era
H OH metabolites OH Review Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era Qimin Hai and Jonathan D. Smith * Department of Cardiovascular & Metabolic Sciences, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-216-444-2248 Abstract: The purification and cloning of the acyl-coenzyme A: cholesterol acyltransferase (ACAT) enzymes and the sterol O-acyltransferase (SOAT) genes has opened new areas of interest in cholesterol metabolism given their profound effects on foam cell biology and intestinal lipid absorption. The generation of mouse models deficient in Soat1 or Soat2 confirmed the importance of their gene products on cholesterol esterification and lipoprotein physiology. Although these studies supported clinical trials which used non-selective ACAT inhibitors, these trials did not report benefits, and one showed an increased risk. Early genetic studies have implicated common variants in both genes with human traits, including lipoprotein levels, coronary artery disease, and Alzheimer’s disease; however, modern genome-wide association studies have not replicated these associations. In contrast, the common SOAT1 variants are most reproducibly associated with testosterone levels. Keywords: cholesterol esterification; atherosclerosis; ACAT; SOAT; inhibitors; clinical trial Citation: Hai, Q.; Smith, J.D. Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its 1. Introduction Discovery to Clinical Trials and the The acyl-coenzyme A:cholesterol acyltransferase (ACAT; EC 2.3.1.26) enzyme family Genomics Era. Metabolites 2021, 11, consists of membrane-spanning proteins, which are primarily located in the endoplasmic 543. https://doi.org/10.3390/ reticulum [1]. -

Bhagwan Moorjani, MD, FAAP, FAAN • Requires Knowledge of Normal CNS Developmental (I.E
1/16/2012 Neuroimaging in Childhood • Neuroimaging issues are distinct from Pediatric Neuroimaging in adults Neurometabolic-degenerative disorder • Sedation/anesthesia and Epilepsy • Motion artifacts Bhagwan Moorjani, MD, FAAP, FAAN • Requires knowledge of normal CNS developmental (i.e. myelin maturation) • Contrast media • Parental anxiety Diagnostic Approach Neuroimaging in Epilepsy • Age of onset • Peak incidence in childhood • Static vs Progressive • Occurs as a co-morbid condition in many – Look for treatable causes pediatric disorders (birth injury, – Do not overlook abuse, Manchausen if all is negative dysmorphism, chromosomal anomalies, • Phenotype presence (syndromic, HC, NCS, developmental delays/regression) systemic involvement) • Predominant symptom (epilepsy, DD, • Many neurologic disorders in children weakness/motor, psychomotor regression, have the same chief complaint cognitive/dementia) 1 1/16/2012 Congenital Malformation • Characterized by their anatomic features • Broad categories: based on embryogenesis – Stage 1: Dorsal Induction: Formation and closure of the neural tube. (Weeks 3-4) – Stage 2: Ventral Induction: Formation of the brain segments and face. (Weeks 5-10) – Stage 3: Migration and Histogenesis: (Months 2-5) – Stage 4: Myelination: (5-15 months; matures by 3 years) Dandy Walker Malformation Dandy walker • Criteria: – high position of tentorium – dysgenesis/agenesis of vermis – cystic dilatation of fourth ventricle • commonly associated features: – hypoplasia of cerebellum – scalloping of inner table of occipital bone • associated abnormalities: – hydrocephalus 75% – dysgenesis of corpus callosum 25% – heterotropia 10% 2 1/16/2012 Etiology of Epilepsy: Developmental and Genetic Classification of Gray Matter Heterotropia Cortical Dysplasia 1. Secondary to abnormal neuronal and • displaced masses of nerve cells • Subependymal glial proliferation/apoptosis (gray matter) heterotropia (most • most common: small nest common) 2. -

Phenotype Heterogeneity of Congenital Adrenal Hyperplasia Due
Kor et al. BMC Medical Genetics (2018) 19:115 https://doi.org/10.1186/s12881-018-0629-2 CASE REPORT Open Access Phenotype heterogeneity of congenital adrenal hyperplasia due to genetic mosaicism and concomitant nephrogenic diabetes insipidus in a sibling Yılmaz Kor1†, Minjing Zou2†, Roua A. Al-Rijjal2, Dorota Monies2, Brian F. Meyer2 and Yufei Shi2* Abstract Background: Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency (21OHD) is an autosomal recessive disorder caused by mutations in the CYP21A2. Congenital nephrogenic diabetes insipidus (NDI) is a rare X- linked recessive or autosomal recessive disorder caused by mutations in either AVPR2 or AQP2. Genotype-phenotype discordance caused by genetic mosaicism in CAH patients has not been reported, nor the concomitant CAH and NDI. Case presentation: We investigated a patient with concomitant CAH and NDI from a consanguineous family. She (S- 1) presented with clitoromegaly at 3 month of age, and polydipsia and polyuria at 13 month of age. Her parents and two elder sisters (S-2 and S-3) were clinically normal, but elevated levels of serum 17-hydroxyprogesterone (17-OHP) were observed in the mother and S-2. The coding region of CYP21A2 and AQP2 were analyzed by PCR-sequencing analysis to identify genetic defects. Two homozygous CYP21A2 mutations (p.R357W and p.P454S) were identified in the proband and her mother and S-2. The apparent genotype-phenotype discordance was due to presence of small amount of wild-type CYP21A2 alleles in S-1, S-2, and their mother’s genome, thus protecting them from development of classic form of 21OHD (C21OHD). -

Association Between Two Polymorphisms in the SRD5A2 Gene and Serum Androgen Concentrations in British Men1
578 Vol. 12, 578–581, June 2003 Cancer Epidemiology, Biomarkers & Prevention Short Communication Association between Two Polymorphisms in the SRD5A2 Gene and Serum Androgen Concentrations in British Men1 Naomi E. Allen,2 Juergen K. V. Reichardt, are needed to additionally clarify the role of the A49T Hannah Nguyen, and Timothy J. Key polymorphism in androgen metabolism. Cancer Research United Kingdom Epidemiology Unit, University of Oxford, Gibson Building, Radcliffe Infirmary, Oxford, OX2 6HE, United Kingdom Introduction [N. E. A., T. J. K.], and Institute for Genetic Medicine, Departments of Biochemistry and Molecular Biology and Preventive Medicine, University of It is well established that the steroid 5 ␣-reductase type II Southern California School of Medicine, University of Southern California, enzyme, which irreversibly converts testosterone to its more Los Angeles, California 90089-9075 [J. K. V. R., H. N.] potent metabolite DHT3 in prostatic cells, is required for the normal growth and development of the prostate gland (1). A meta-analysis of prospective studies suggests that the serum Abstract concentration of A-diol-g, a serum marker of the abundance of Androgens are essential for the growth of the prostate 5 ␣-reductase type II and intraprostatic DHT, is slightly higher gland and have been implicated in the development of in men who subsequently develop prostate cancer compared prostate cancer. Little is known about the determinants with healthy individuals (2). The circulating A-diol-g concen- of androgen levels in men, although observed ethnic tration is believed to be a more accurate serum marker of 5 differences suggest they may have a genetic basis. -

Diagnosis, Treatment and Follow Up
DOI: 10.1002/jimd.12024 REVIEW International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow up Ruqaiah Altassan1,2 | Romain Péanne3,4 | Jaak Jaeken3 | Rita Barone5 | Muad Bidet6 | Delphine Borgel7 | Sandra Brasil8,9 | David Cassiman10 | Anna Cechova11 | David Coman12,13 | Javier Corral14 | Joana Correia15 | María Eugenia de la Morena-Barrio16 | Pascale de Lonlay17 | Vanessa Dos Reis8 | Carlos R Ferreira18,19 | Agata Fiumara5 | Rita Francisco8,9,20 | Hudson Freeze21 | Simone Funke22 | Thatjana Gardeitchik23 | Matthijs Gert4,24 | Muriel Girad25,26 | Marisa Giros27 | Stephanie Grünewald28 | Trinidad Hernández-Caselles29 | Tomas Honzik11 | Marlen Hutter30 | Donna Krasnewich18 | Christina Lam31,32 | Joy Lee33 | Dirk Lefeber23 | Dorinda Marques-da-Silva9,20 | Antonio F Martinez34 | Hossein Moravej35 | Katrin Õunap36,37 | Carlota Pascoal8,9 | Tiffany Pascreau38 | Marc Patterson39,40,41 | Dulce Quelhas14,42 | Kimiyo Raymond43 | Peymaneh Sarkhail44 | Manuel Schiff45 | Małgorzata Seroczynska29 | Mercedes Serrano46 | Nathalie Seta47 | Jolanta Sykut-Cegielska48 | Christian Thiel30 | Federic Tort27 | Mari-Anne Vals49 | Paula Videira20 | Peter Witters50,51 | Renate Zeevaert52 | Eva Morava53,54 1Department of Medical Genetic, Montréal Children's Hospital, Montréal, Québec, Canada 2Department of Medical Genetic, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia 3Department of Human Genetics, KU Leuven, Leuven, Belgium 4LIA GLYCOLAB4CDG (International -

Advances in Understanding the Genetics of Syndromes Involving Congenital Upper Limb Anomalies
Review Article Page 1 of 10 Advances in understanding the genetics of syndromes involving congenital upper limb anomalies Liying Sun1#, Yingzhao Huang2,3,4#, Sen Zhao2,3,4, Wenyao Zhong1, Mao Lin2,3,4, Yang Guo1, Yuehan Yin1, Nan Wu2,3,4, Zhihong Wu2,3,5, Wen Tian1 1Hand Surgery Department, Beijing Jishuitan Hospital, Beijing 100035, China; 2Beijing Key Laboratory for Genetic Research of Skeletal Deformity, Beijing 100730, China; 3Medical Research Center of Orthopedics, Chinese Academy of Medical Sciences, Beijing 100730, China; 4Department of Orthopedic Surgery, 5Department of Central Laboratory, Peking Union Medical College Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing 100730, China Contributions: (I) Conception and design: W Tian, N Wu, Z Wu, S Zhong; (II) Administrative support: All authors; (III) Provision of study materials or patients: All authors; (IV) Collection and assembly of data: Y Huang; (V) Data analysis and interpretation: L Sun; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Wen Tian. Hand Surgery Department, Beijing Jishuitan Hospital, Beijing 100035, China. Email: [email protected]. Abstract: Congenital upper limb anomalies (CULA) are a common birth defect and a significant portion of complicated syndromic anomalies have upper limb involvement. Mostly the mortality of babies with CULA can be attributed to associated anomalies. The cause of the majority of syndromic CULA was unknown until recently. Advances in genetic and genomic technologies have unraveled the genetic basis of many syndromes- associated CULA, while at the same time highlighting the extreme heterogeneity in CULA genetics. Discoveries regarding biological pathways and syndromic CULA provide insights into the limb development and bring a better understanding of the pathogenesis of CULA. -

WAGR Syndrome: Clinical Features and Guidelines for Management
WAGR syndrome: clinical features and guidelines for management Kelly Trout, BSN, RN International WAGR Syndrome Association 2020 Introduction WAGR syndrome is a rare multiple congenital-anomaly syndrome caused by interstitial deletion of the distal portion of chromosome 11p13. Deletion size varies between individuals from 1 million to 26.5 million base pairs, with an average of 11 million base pairs. Variability in size of the genetic deletion is thought to account for the variable phenotype. WAGR is an acronym for the most prominent features: W is for Wilms tumor, A for aniridia, G for genitourinary anomalies, and R for range of developmental delays. Wilms tumor and genital anomalies are caused by deletion of the WT1 tumor-suppressor gene, and aniridia is caused by deletion of the PAX6 ocular development gene. Developmental delays are presumed to be the result of deletion of as yet unidentified genes in the region. Most cases are identified by chromosome studies of children with isolated aniridia and are due to de novo deletions, although a few familial translocations have been reported. Individuals with WAGR syndrome have a high risk for development of Wilms tumor and late-onset renal failure, as well as a variety of additional associated conditions. Diagnosis ● Most cases of WAGR syndrome are identified in infants with isolated aniridia, 30% of whom will be positive for the characteristic deletion (11p13) ● In rare cases, aniridia may not be present. Children with Wilms tumor and genital anomalies may also warrant genetic testing ● -
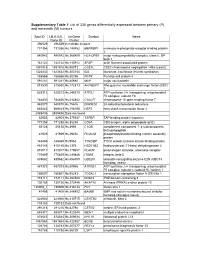
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

Diagnosis of Abnormalities in Gonadal Development BERNARD GONDOS, M.D
ANNALS OF CLINICAL AND LABORATORY SCIENCE, Vol. 12, No. 4 Copyright © 1982, Institute for Clinical Science, Inc. Diagnosis of Abnormalities in Gonadal Development BERNARD GONDOS, M.D. Department of Pathology, University of Connecticut, Farmington, CT 06032 ABSTRACT The role of the clinical laboratory in the diagnosis of abnormalities in gonadal development is reviewed, beginning with a description of the normal differentiation of the ovary and testis and the major types of disorders encountered. The conditions are classified as resulting from abnormal go nadal differentiation, defective endocrine function or excessive endocrine activity. Germ cell neoplasms are also reviewed. Laboratory procedures utilized in evaluation of gonadal abnormalities include cytogenetic, hor monal, and histopathologic studies. Standard procedures are described as well as newer methods which have undergone increasing use in recent years and other specialized procedures which are under investigation for possible clinical application. Introduction tors may all play a role in the development of structural and functional abnormalities The role of the laboratory in the diagno of gonadal differentiation. As a result, sis of abnormalities in gonadal develop classifications of intersex disorders and ment is particularly important. Because of abnormalities of hormone production are the many varieties of such disorders and often confusing. their complex pathogenesis, the types of The present report reviews the labora laboratory tests utilized are quite varied. tory diagnosis of disorders of gonadal de The applications and significance of these velopment, beginning with a consider tests should be clearly understood, since ation of normal gonadal differentiation proper utilization and evaluation may be and a brief summary of the main categor critical in determining gender role assign ies of abnormalities. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Evidence for Cross Species Extrapolation of Mammalian-Based High-Throughput Screening Assay Results † † ‡ † Carlie A
Article Cite This: Environ. Sci. Technol. 2018, 52, 13960−13971 pubs.acs.org/est Evidence for Cross Species Extrapolation of Mammalian-Based High-Throughput Screening Assay Results † † ‡ † Carlie A. LaLone,*, Daniel L. Villeneuve, Jon A. Doering, Brett R. Blackwell, § § ¶ † || Thomas R. Transue, Cody W. Simmons, Joe Swintek, Sigmund J. Degitz, Antony J. Williams, † and Gerald T. Ankley † Office of Research and Development, National Health and Environmental Effects Research Laboratory, Mid-Continent Ecology Division, US Environmental Protection Agency, 6201 Congdon Blvd., Duluth, Minnesota 55804, United States ‡ National Research Council, 6201 Congdon Blvd., Duluth, Minnesota 55804, United States § CSRA Inc., 109 T.W. Alexander Drive, Research Triangle Park, North Carolina 27711, United States ¶ Badger Technical Services, 6201 Congdon Blvd., Duluth, Minnesota 55804, United States || Office of Research and Development, National Center for Computational Toxicology, US Environmental Protection Agency, 109 T.W. Alexander Drive, Research Triangle Park, North Carolina 27711, United States *S Supporting Information ABSTRACT: High-throughput screening (HTS) and computational technologies have emerged as important tools for chemical hazard identification. The US Environmental Protection Agency (EPA) launched the Toxicity ForeCaster (ToxCast) Program, which has screened thousands of chemicals in hundreds of mammalian-based HTS assays for biological activity. The data are being used to prioritize toxicity testing on those chemi- cals likely to lead to adverse effects. To use HTS assays in predicting hazard to both humans and wildlife, it is necessary to understand how broadly these data may be extrapolated across species. The US EPA Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS; https://seqapass.epa.gov/seqapass/) tool was used to assess conservation of the 484 protein targets represented in the suite of ToxCast assays and other HTS assays.