Modern Methods of Organic Synthesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -

Xerox University Microfilms, Ann Arbor, M Ichigan 48106
I I I 77-2394 EDWARDS, Robert Charles, 1949- SYNTHESES AND CHARACTERIZATION OF CHROMIUM COMPLEXES WITH TETRAAZA MACROCYCLIC LIGANDS. The Ohio State University, Ph.D., 1976 Chemistry, inorganic Xerox University Microfilms, Ann Arbor, Michigan 48106 SYNTHESES AND CHARACTERIZATION OF CHROMIUM COMPLEXES WITH TETRAAZA MACROCYCLIC LIGANDS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Robert Charles Edwards, B.S. The Ohio State University 1976 Reading Committee: Approved by Professor Daryle H. Busch Professor Devon W. Meek Professor Eugene P. Schram Advised Department of Chemistry To Debra and Marie & # 5J: aj: % # # # ACKNOWLEDGEMENTS I would like to acknowledge the help given to me by fellow graduate students, post-doctoral fellows in Dr. Busch's group, and the staff in the Department of Chemistry. I want to especially thank Professor Daryle H„ Busch for his guidance and understanding,, iii CURRICULUM VITAE March 2, 1949. ................ ... ..................................................... Born, Marion, Ohio 1971 .................................................. ................................................ B .S., Heidelberg College Tiffin, Ohio 1971-1974. ....................................................................................... Teaching Associate, Dept, of Chemistry, The Ohio State University, Columbus, Ohio 1974-197 5 ................................................................................. Allied-Chemical -

Catalytic Pyrolysis of Plastic Wastes for the Production of Liquid Fuels for Engines
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2019 Supporting information for: Catalytic pyrolysis of plastic wastes for the production of liquid fuels for engines Supattra Budsaereechaia, Andrew J. Huntb and Yuvarat Ngernyen*a aDepartment of Chemical Engineering, Faculty of Engineering, Khon Kaen University, Khon Kaen, 40002, Thailand. E-mail:[email protected] bMaterials Chemistry Research Center, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen, 40002, Thailand Fig. S1 The process for pelletization of catalyst PS PS+bentonite PP ) t e PP+bentonite s f f o % ( LDPE e c n a t t LDPE+bentonite s i m s n HDPE a r T HDPE+bentonite Gasohol 91 Diesel 4000 3500 3000 2500 2000 1500 1000 500 Wavenumber (cm-1) Fig. S2 FTIR spectra of oil from pyrolysis of plastic waste type. Table S1 Compounds in oils (%Area) from the pyrolysis of plastic wastes as detected by GCMS analysis PS PP LDPE HDPE Gasohol 91 Diesel Compound NC C Compound NC C Compound NC C Compound NC C 1- 0 0.15 Pentane 1.13 1.29 n-Hexane 0.71 0.73 n-Hexane 0.65 0.64 Butane, 2- Octane : 0.32 Tetradecene methyl- : 2.60 Toluene 7.93 7.56 Cyclohexane 2.28 2.51 1-Hexene 1.05 1.10 1-Hexene 1.15 1.16 Pentane : 1.95 Nonane : 0.83 Ethylbenzen 15.07 11.29 Heptane, 4- 1.81 1.68 Heptane 1.26 1.35 Heptane 1.22 1.23 Butane, 2,2- Decane : 1.34 e methyl- dimethyl- : 0.47 1-Tridecene 0 0.14 2,2-Dimethyl- 0.63 0 1-Heptene 1.37 1.46 1-Heptene 1.32 1.35 Pentane, -

Title Crystallization of Stereospecific Olefin Copolymers (Special Issue on Physical Chemistry) Author(S) Sakaguchi, Fumio; Kita
Crystallization of Stereospecific Olefin Copolymers (Special Title Issue on Physical Chemistry) Author(s) Sakaguchi, Fumio; Kitamaru, Ryozo; Tsuji, Waichiro Bulletin of the Institute for Chemical Research, Kyoto Citation University (1966), 44(4): 295-315 Issue Date 1966-10-31 URL http://hdl.handle.net/2433/76134 Right Type Departmental Bulletin Paper Textversion publisher Kyoto University Crystallization of Stereospecifie Olefin Copolymers Fumio SAKAGUCHI,Ryozo KITAMARU and Waichiro TSUJI* (Tsuji Laboratory) Received August 13, 1966 The stereoregularity of isotactic poly(4-methyl-1-pentene) was characterized and isomorphism phenomena were examined for the copolymeric systems of 4-methyl-1-pentene with several olefins in order to study the crystallization phenomena in these olefin copoly- mers polymerized with stereospecific catalysts. The structural heterogeneity or the fine crystalline structure of poly(4-methyl-1-pentene) could be correlated with its molecular structure by viewing this stereoregular homopolymer as if it were a copolymer. Cocrystallization or isomorphism phenomenon was recognized for the copolymeric systems of 4-methyl-1-pentene with butene-1, pentene-1, decene-1 and 3-methyl-1-butene, while no evidence of the phenomenon was obtained for the copolymeric systems with styrene and propylene. The degree of the isomorphism of those copolymers was discussed with the informations on the crystalline phases obtained from the X-ray study, on the constitution of the copolymeric chains in the amorphous phases obtained from the viscoelastic studies and on the other thermodynamical properties of these systems. INTRODUCTION Many works have been made with regard to the homopolymerization of olefins with stereospecific catalysts, i. e. complex catalysts composed of the combination of organometallic compound and transitional metallic compound. -

© Copyright 2013 Jennifer L. Steele
© Copyright 2013 Jennifer L. Steele DIVALENT TRANSITION METAL CENTERS: THE SYNTHESIS OF NEW CHEMICAL VAPOR DEPOSITION PRECURSORS AND STUDIES OF ETHYLENE POLYMERIZATION AND OLIGOMERIZATION CATALYSTS BY JENNIFER L. STEELE DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate College of the University of Illinois at Urbana-Champaign, 2013 Urbana, Illinois Doctoral Committee: Professor Gregory S. Girolami, Chair Assistant Professor Alison R. Fout Professor John A. Katzenellenbogen Professor Thomas B. Rauchfuss Abstract Volatile transition metal complexes that contain boron hydride ligands are desirable for their potential as precursors for metal diboride films for microelectronics applications. Recently our group has discovered a new class of potential precursors in the metal complexes of the chelating borohydride, N,N-dimethylaminodiboranate (DMADB). To date, attempts to synthesize homoleptic complexes of the late transition metals have afforded intractable mixtures, likely the result of overreduction of the metal center. This work has focused on the synthesis and characterization of heteroleptic complexes of the late transition metals that contain both DMADB and 1,2,3,4,5,-pentamethylcyclopentadienyl ligands. The reaction of metal complexes of the form [Cp*MX]n, where Cp* is 1,2,3,4,5,- pentamethylcyclopentadienyl, M = Cr, Fe, Co, or Ru, and X = Cl or I with sodium dimethylaminodiboranate (NaDMADB) in diethyl ether affords the divalent complexes [Cp*M(DMADB)]. Additionally, the analogous vanadium compound [Cp*V(DMADB)] can be synthesized from the reduction of [Cp*VCl2]3 with NaDMADB in diethyl ether. All of these compounds are volatile under static vacuum at room temperature, but are also thermally sensitive; the iron and ruthenium derivatives decompose at room temperature over a day. -

Syntheses and Eliminations of Cyclopentyl Derivatives David John Rausch Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1966 Syntheses and eliminations of cyclopentyl derivatives David John Rausch Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Rausch, David John, "Syntheses and eliminations of cyclopentyl derivatives " (1966). Retrospective Theses and Dissertations. 2875. https://lib.dr.iastate.edu/rtd/2875 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 66—6996 RAUSCH, David John, 1940- SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES. Iowa State University of Science and Technology Ph.D., 1966 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES by David John Rausch A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Organic Chemistry Approved : Signature was redacted for privacy. Signature was redacted for privacy. Head of Major Department Signature was redacted for privacy. Iowa State University Of Science and Technology Ames, Iowa 1966 ii TABLE OF CONTENTS VITA INTRODUCTION HISTORICAL Conformation of Cyclopentanes Elimination Reactions RESULTS AND DISCUSSION Synthetic Elimination Reactions EXPERIMENTAL Preparation and Purification of Materials Procedures and Data for Beta Elimination Reactions SUMMARY LITERATURE CITED ACKNOWLEDGEMENTS iii VITA The author was born in Aurora, Illinois, on October 24, 1940, to Mr. -

Catalytic Asymmetric Di Hydroxylation
Chem. Rev. 1994, 94, 2483-2547 2483 Catalytic Asymmetric Dihydroxylation Hartmuth C. Kolb,t Michael S. VanNieuwenhze,* and K. Barry Sharpless* Department of Chemistry, The Scripps Research Institute, 10666 North Torfey Pines Road, La Jolla, California 92037 Received Ju/y 28, 1994 (Revised Manuscript Received September 14, 1994) Contents 3.1.4. Differentiation of the Hydroxyl Groups by 2524 Selective, Intramolecular Trapping 1. Introduction and General Principles 2483 3.1.5. Miscellaneous Transformations 2525 2. Enantioselective Preparation of Chiral 1,2-Diols 2489 3.2. Preparation of Chiral Building Blocks 2527 from Olefins 3.2.1. Electrophilic Building Blocks 2527 2.1. Preparation of Ligands, Choice of Ligand, 2489 Scope, and Limitations 3.2.2. Chiral Diol and Polyol Building Blocks 2529 2.1.1. Preparation of the Ligands 2490 3.2.3. Chiral Monohydroxy Compounds Derived 2529 from Diols 2.1 -2. Ligand Choice and Enantioselectivity Data 2490 3.2.4. 5- and 6-Membered Heterocycles 2530 2.1.3. Limitations 2491 3.3. Preparation of Chiral Auxiliaries for Other 2530 2.2. Reaction Conditions 2493 Asymmetric Transformations 2.2.1. Asymmetric Dihydroxylation of the 2493 3.3.1. Preparation of 2530 “Standard Substrates” (1 R,2S)-trans-2-phenylcyclohexanol 2.2.2. Asymmetric Dihydroxylation of 2496 3.3.2. Optically Pure Hydrobenzoin (Stilbenediol) 2531 Tetrasubstituted Olefins, Including Enol and Derivatives Ethers 4. Recent Applications: A Case Study 2536 2.2.3. Asymmetric Dihydroxylation of 2496 Electron-Deficient Olefins 5. Conclusion 2538 2.2.4. Chemoselectivity in the AD of Olefins 2497 6. References and Footnotes 2542 Containing Sulfur 2.3. -

Conformational Studies of Aliphatic Secondary Ozonides (Propene, 1-Butene and 1-Heptene) by Means of FTIR Spectroscopy
DOI: 10.2478/s11532-006-0031-3 Research article CEJC 4(4) 2006 578–591 Conformational studies of aliphatic secondary ozonides (propene, 1-butene and 1-heptene) by means of FTIR spectroscopy Ruta Bariseviciute, Justinas Ceponkus, Alytis Gruodis, Valdas Sablinskas∗ Department of General Physics and Spectroscopy, Vilnius University, Vilnius-01513, Lithuania Received 6 January 2006; accepted 31 May 2006 Abstract: Ozonization reaction of simple alkenes was studied by means of FT infrared absorption gas spectroscopy. The reaction was performed at 95 K in neat films of the reactants. IR absorption spectra of the gaseous products were recorded. The spectra were analyzed combining experimental results with theoretical calculations performed at B3LYP 6-311++G (3df, 3pd) level. We found that among all theoretically predicted conformers of propene secondary ozonide, only one which has the O-O half-chair configuration for the five membered ring and the radical attached in the equatorial position was present in the sample. Samples of 1-butene and 1-heptene secondary ozonides consist from two conformers of very similar energy (ΔH =0.3 kJ/mol). The most stable conformer for both ozonides is the one with O-O half-chair configuration of the five membered ring and the radical attached in equatorial position and the aliphatic chain in gauche position. The second stable conformer has the aliphatic chain in anti position. c Versita Warsaw and Springer-Verlag Berlin Heidelberg. All rights reserved. Keywords: Ozonization, conformational analysis, secondary ozonides, IR spectroscopy 1 Introduction The ability of ozone to cleave double bonds in olefins has been known for more than 50 years. -

Arenechromium Tricarbonyl Complexes: Conformational
η6 – ARENECHROMIUM TRICARBONYL COMPLEXES: CONFORMATIONAL ANALYSIS, STEREOCONTROL IN NUCLEOPHILIC ADDITION AND APPLICATIONS IN ORGANIC SYNTHESIS by HARINANDINI PARAMAHAMSAN Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Thesis Advisor: Prof. Anthony J. Pearson Department of Chemistry CASE WESTERN RESERVE UNIVERSITY May 2005 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the dissertation of Harinandini Paramahamsan candidate for the Ph.D. degree*. (signed) Prof. Philip P. Garner (Chair of the Committee, Department of Chemistry, CWRU) Prof. Anthony J. Pearson (Department of Chemistry, CWRU) Prof. Fred L. Urbach (Department of Chemistry, CWRU) Dr. Zwong-Wu Guo (Department of Chemistry, CWRU) Dr. Stuart J. Rowan (Department of Macromolecular Science and Engineering, CWRU) Date: 14th January 2005 *We also certify that written approval has been obtained for any propriety material contained therein. To Amma, Naina & all my Teachers Table of Contents List of Tables………………………………………………………………………..……iv List of Figures…………………………………………………………………….…........vi List of Schemes…………………………………………………………………….….….ix List of Equations………………………………………………………...……….……….xi Acknowledgements………………………………………………………….…..……….xii List of Abbreviations……………………………………………………………………xiv Abstract………………………………………………………………………………….xvi CHAPTER I........................................................................................................................ 1 I.1 Structure and Bonding ........................................................................................... -

Properties and Synthesis. Elimination Reactions of Alkyl Halides
P1: PBU/OVY P2: PBU/OVY QC: PBU/OVY T1: PBU Printer: Bind Rite JWCL234-07 JWCL234-Solomons-v1 December 8, 2009 21:37 7 ALKENES AND ALKYNES I: PROPERTIES AND SYNTHESIS. ELIMINATION REACTIONS OF ALKYL HALIDES SOLUTIONS TO PROBLEMS 7.1 (a) ( E )-1-Bromo-1-chloro-1-pentene or ( E )-1-Bromo-1-chloropent-1-ene (b) ( E )-2-Bromo-1-chloro-1-iodo-1-butene or ( E )-2-Bromo-1-chloro-1-iodobut-1-ene (c) ( Z )-3,5-Dimethyl-2-hexene or ( Z )-3,5-Dimethylhex-2-ene (d) ( Z )-1-Chloro-1-iodo-2-methyl-1-butene or ( Z )-1-Chloro-1-iodo-2-methylbut-1-ene (e) ( Z,4 S )-3,4-Dimethyl-2-hexene or ( Z,4 S )-3,4-Dimethylhex-2-ene (f) ( Z,3 S )-1-Bromo-2-chloro-3-methyl-1-hexene or (Z,3 S )-1-Bromo-2-chloro-3-methylhex-1-ene 7.2 < < Order of increasing stability 7.3 (a), (b) H − 2 ∆ H° = − 119 kJ mol 1 Pt 2-Methyl-1-butene pressure (disubstituted) H2 − ∆ H° = − 127 kJ mol 1 Pt 3-Methyl-1-butene pressure (monosubstituted) H2 − ∆ H° = − 113 kJ mol 1 Pt 2-Methyl-2-butene pressure (trisubstituted) (c) Yes, because hydrogenation converts each alkene into the same product. 106 CONFIRMING PAGES P1: PBU/OVY P2: PBU/OVY QC: PBU/OVY T1: PBU Printer: Bind Rite JWCL234-07 JWCL234-Solomons-v1 December 8, 2009 21:37 ALKENES AND ALKYNES I: PROPERTIES AND SYNTHESIS 107 H H (d) > > H H H (trisubstituted) (disubstituted) (monosubstituted) Notice that this predicted order of stability is confirmed by the heats of hydro- genation. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -
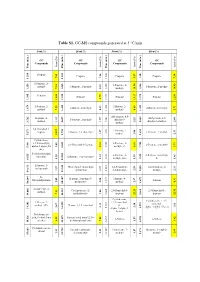
Table S1. GC-MS Compounds Generated at 5 °C/Min
Table S1. GC-MS compounds generated at 5 °C/min. 0 wt.% 10 wt.% 30 wt.% 50 wt.% GC GC GC GC Compounds Compounds Compounds Compounds Area (%) Area (%) Area (%) Area (%) Time (min.) (min.) Time Time (min.) (min.) Time (min.) Time (min.) Time Propene Propene Propene Propene 7.42 7.42 1.26 5.47 1.20 7.38 1.200 1.200 1.200 1.214 1.214 1-Propene, 2- 1-Propene, 2- methyl- 1-Propene, 2-methyl- 1-Propene, 2-methyl- 7.34 7.34 2.41 methyl- 1.245 1.245 1.245 1.259 1.259 10.54 1.246 14.90 Pentane Pentane Pentane Pentane 3.79 3.79 1.375 1.375 1.395 1.395 11.81 1.381 13.17 1.375 12.83 1-Pentene, 2- 2-Butene, 2- 2-Butene, 2-methyl- 2-Butene, 2-methyl- methyl- 7.25 0.93 methyl- 4.65 5.71 1.426 1.426 1.420 1.420 1.679 1.679 1H-Pyrazole, 4,5- Heptane, 4- 1H-Pyrazole, 4,5- 1-Pentene, 2-methyl- dihydro-5- methyl- 1.92 1.57 2.25 dihydro-5-methyl- 2.44 1.659 1.659 1.562 1.569 3.762 3.762 methyl- 2,4-Dimethyl-1- 1-Pentene, 2- heptene 2-Butene, 2,3-dimethyl- 1-Pentene, 2-methyl- 1.27 1.27 methyl- 3.08 2.33 1.653 1.653 1.653 5.671 5.671 34.57 1.750 Cyclohexane, 1,3,5-trimethyl-, 2-Pentene, 3- 2,4-Dimethyl-1-heptene 2-Pentene, 2-methyl- alpha.,3.alpha.,5.b 2.03 9.11 methyl-, €- 6.11 5.73 5.605 5.605 1.743 1.744 5.981 5.981 eta.)- 5-Aminoisoxazole 2-Pentene, 3- 2-Pentene, 3-methyl-, Spermine Ethanone, 1-cyclopentyl- 1.55 1.55 1.32 methyl-, (Z)- 1.59 €- 1.46 1.828 1.828 1.828 6.925 6.925 10.560 Ethanone, 1- 1R,2c,3t,4t-Tetramethyl- 1,3-Pentadiene, 1,4-Hexadiene, 5- cyclopentyl- 3.57 3.57 cyclohexan 0.97 2,3-dimethyl- 2.28 methyl- 2.31 2.584 2.584 2.585 10.613 10.613 10.638 N- Heptane, 2-methyl-3- Heptane, 4- Methylallylamine Toluene 2.49 2.49 methylene- 2.50 methyl- 2.97 4.31 3.697 3.697 3.697 14.047 14.047 10.684 10.684 2-Undecene, 4- Cyclopentane, (2- 2,4-Dimethyl-1- 2,4-Dimethyl-1- methyl- 7.81 7.81 methylbutyl)- 1.00 heptene heptene 5.605 5.605 22.29 22.29 5.605 12.95 14.086 14.086 14.163 Cyclohexane, Cyclohexane, 1,3,5- 2-Decene, 7- 1,3,5-trimethyl-, trimethyl- methyl-, (Z)- Hexane, 2,3,4-trimethyl- (1.