An Improved Method for Utilizing High‐Throughput Amplicon Sequencing to Determine the Diets of Insectivorous Animals
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ARTHROPOD COMMUNITIES and PASSERINE DIET: EFFECTS of SHRUB EXPANSION in WESTERN ALASKA by Molly Tankersley Mcdermott, B.A./B.S
Arthropod communities and passerine diet: effects of shrub expansion in Western Alaska Item Type Thesis Authors McDermott, Molly Tankersley Download date 26/09/2021 06:13:39 Link to Item http://hdl.handle.net/11122/7893 ARTHROPOD COMMUNITIES AND PASSERINE DIET: EFFECTS OF SHRUB EXPANSION IN WESTERN ALASKA By Molly Tankersley McDermott, B.A./B.S. A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science in Biological Sciences University of Alaska Fairbanks August 2017 APPROVED: Pat Doak, Committee Chair Greg Breed, Committee Member Colleen Handel, Committee Member Christa Mulder, Committee Member Kris Hundertmark, Chair Department o f Biology and Wildlife Paul Layer, Dean College o f Natural Science and Mathematics Michael Castellini, Dean of the Graduate School ABSTRACT Across the Arctic, taller woody shrubs, particularly willow (Salix spp.), birch (Betula spp.), and alder (Alnus spp.), have been expanding rapidly onto tundra. Changes in vegetation structure can alter the physical habitat structure, thermal environment, and food available to arthropods, which play an important role in the structure and functioning of Arctic ecosystems. Not only do they provide key ecosystem services such as pollination and nutrient cycling, they are an essential food source for migratory birds. In this study I examined the relationships between the abundance, diversity, and community composition of arthropods and the height and cover of several shrub species across a tundra-shrub gradient in northwestern Alaska. To characterize nestling diet of common passerines that occupy this gradient, I used next-generation sequencing of fecal matter. Willow cover was strongly and consistently associated with abundance and biomass of arthropods and significant shifts in arthropod community composition and diversity. -

Field Release of the Leaf-Feeding Moth, Hypena Opulenta (Christoph)
United States Department of Field release of the leaf-feeding Agriculture moth, Hypena opulenta Marketing and Regulatory (Christoph) (Lepidoptera: Programs Noctuidae), for classical Animal and Plant Health Inspection biological control of swallow- Service worts, Vincetoxicum nigrum (L.) Moench and V. rossicum (Kleopow) Barbarich (Gentianales: Apocynaceae), in the contiguous United States. Final Environmental Assessment, August 2017 Field release of the leaf-feeding moth, Hypena opulenta (Christoph) (Lepidoptera: Noctuidae), for classical biological control of swallow-worts, Vincetoxicum nigrum (L.) Moench and V. rossicum (Kleopow) Barbarich (Gentianales: Apocynaceae), in the contiguous United States. Final Environmental Assessment, August 2017 Agency Contact: Colin D. Stewart, Assistant Director Pests, Pathogens, and Biocontrol Permits Plant Protection and Quarantine Animal and Plant Health Inspection Service U.S. Department of Agriculture 4700 River Rd., Unit 133 Riverdale, MD 20737 Non-Discrimination Policy The U.S. Department of Agriculture (USDA) prohibits discrimination against its customers, employees, and applicants for employment on the bases of race, color, national origin, age, disability, sex, gender identity, religion, reprisal, and where applicable, political beliefs, marital status, familial or parental status, sexual orientation, or all or part of an individual's income is derived from any public assistance program, or protected genetic information in employment or in any program or activity conducted or funded by the Department. (Not all prohibited bases will apply to all programs and/or employment activities.) To File an Employment Complaint If you wish to file an employment complaint, you must contact your agency's EEO Counselor (PDF) within 45 days of the date of the alleged discriminatory act, event, or in the case of a personnel action. -

Water Ferns Azolla Spp. (Azollaceae) As New Host Plants for the Small China-Mark Moth, Cataclysta Lemnata (Linnaeus, 1758) (Lepidoptera, Crambidae, Acentropinae)
©Societas Europaea Lepidopterologica; download unter http://www.soceurlep.eu/ und www.zobodat.at Nota Lepi. 40(1) 2017: 1–13 | DOI 10.3897/nl.40.10062 Water ferns Azolla spp. (Azollaceae) as new host plants for the small China-mark moth, Cataclysta lemnata (Linnaeus, 1758) (Lepidoptera, Crambidae, Acentropinae) Atousa Farahpour-Haghani1,2, Mahdi Hassanpour1, Faramarz Alinia2, Gadir Nouri-Ganbalani1, Jabraeil Razmjou1, David Agassiz3 1 University of Mohaghegh Ardabili, Faculty of Agriculture and Natural Resources, Department of Plant Protection, Ardabil, Iran 2 Rice Research Institute of Iran (RRII), Agricultural Research, Education and Extension Organization (AREEO), Rasht, Iran 3 Department of Life Sciences, Natural History Museum, London SW7 5BD, England http://zoobank.org/307196B8-BB55-492B-8ECC-1F518D9EC9E4 Received 1 August 2016; accepted 3 November 2016; published: 20 January 2017 Subject Editor: Bernard Landry. Abstract. Water ferns (Azolla spp., Azollaceae) are reported for the first time as host plants for the larvae of the small China-mark moth Cataclysta lemnata (Linnaeus) (Lepidoptera: Crambidae: Acentropinae) in rice fields and waterways of northern Iran. Cataclysta lemnata is a semi-aquatic species that has been recorded to feed on Lemnaceae and a few other aquatic plants. However, it has not been reported before on Azolla spp. Larvae use water fern as food source and shelter and, at high population density in the laboratory, they completely wiped water fern from the water surface. Feeding was confirmed after rearing more than eight continual generations of C. lemnata on water fern in the laboratory. Adults obtained this way are darker and have darker fuscous markings in both sexes compared with specimens previously reported and the pattern remains unchanged after several generations. -

1 Appendix 3. Thousand Islands National Park Taxonomy Report
Appendix 3. Thousand Islands National Park Taxonomy Report Class Order Family Genus Species Arachnida Araneae Agelenidae Agelenopsis Agelenopsis potteri Agelenopsis utahana Anyphaenidae Anyphaena Anyphaena celer Hibana Hibana gracilis Araneidae Araneus Araneus bicentenarius Larinioides Larinioides cornutus Larinioides patagiatus Clubionidae Clubiona Clubiona abboti Clubiona bishopi Clubiona canadensis Clubiona kastoni Clubiona obesa Clubiona pygmaea Elaver Elaver excepta Corinnidae Castianeira Castianeira cingulata Phrurolithus Phrurolithus festivus Dictynidae Emblyna Emblyna cruciata Emblyna sublata Eutichuridae Strotarchus Strotarchus piscatorius Gnaphosidae Herpyllus Herpyllus ecclesiasticus Zelotes Zelotes hentzi Linyphiidae Ceraticelus Ceraticelus atriceps 1 Collinsia Collinsia plumosa Erigone Erigone atra Hypselistes Hypselistes florens Microlinyphia Microlinyphia mandibulata Neriene Neriene radiata Soulgas Soulgas corticarius Spirembolus Lycosidae Pardosa Pardosa milvina Pardosa moesta Piratula Piratula canadensis Mimetidae Mimetus Mimetus notius Philodromidae Philodromus Philodromus peninsulanus Philodromus rufus vibrans Philodromus validus Philodromus vulgaris Thanatus Thanatus striatus Phrurolithidae Phrurotimpus Phrurotimpus borealis Pisauridae Dolomedes Dolomedes tenebrosus Dolomedes triton Pisaurina Pisaurina mira Salticidae Eris Eris militaris Hentzia Hentzia mitrata Naphrys Naphrys pulex Pelegrina Pelegrina proterva Tetragnathidae Tetragnatha 2 Tetragnatha caudata Tetragnatha shoshone Tetragnatha straminea Tetragnatha viridis -

Lepidoptera of North America 5
Lepidoptera of North America 5. Contributions to the Knowledge of Southern West Virginia Lepidoptera Contributions of the C.P. Gillette Museum of Arthropod Diversity Colorado State University Lepidoptera of North America 5. Contributions to the Knowledge of Southern West Virginia Lepidoptera by Valerio Albu, 1411 E. Sweetbriar Drive Fresno, CA 93720 and Eric Metzler, 1241 Kildale Square North Columbus, OH 43229 April 30, 2004 Contributions of the C.P. Gillette Museum of Arthropod Diversity Colorado State University Cover illustration: Blueberry Sphinx (Paonias astylus (Drury)], an eastern endemic. Photo by Valeriu Albu. ISBN 1084-8819 This publication and others in the series may be ordered from the C.P. Gillette Museum of Arthropod Diversity, Department of Bioagricultural Sciences and Pest Management Colorado State University, Fort Collins, CO 80523 Abstract A list of 1531 species ofLepidoptera is presented, collected over 15 years (1988 to 2002), in eleven southern West Virginia counties. A variety of collecting methods was used, including netting, light attracting, light trapping and pheromone trapping. The specimens were identified by the currently available pictorial sources and determination keys. Many were also sent to specialists for confirmation or identification. The majority of the data was from Kanawha County, reflecting the area of more intensive sampling effort by the senior author. This imbalance of data between Kanawha County and other counties should even out with further sampling of the area. Key Words: Appalachian Mountains, -

Micro-Moth Grading Guidelines (Scotland) Abhnumber Code
Micro-moth Grading Guidelines (Scotland) Scottish Adult Mine Case ABHNumber Code Species Vernacular List Grade Grade Grade Comment 1.001 1 Micropterix tunbergella 1 1.002 2 Micropterix mansuetella Yes 1 1.003 3 Micropterix aureatella Yes 1 1.004 4 Micropterix aruncella Yes 2 1.005 5 Micropterix calthella Yes 2 2.001 6 Dyseriocrania subpurpurella Yes 2 A Confusion with fly mines 2.002 7 Paracrania chrysolepidella 3 A 2.003 8 Eriocrania unimaculella Yes 2 R Easier if larva present 2.004 9 Eriocrania sparrmannella Yes 2 A 2.005 10 Eriocrania salopiella Yes 2 R Easier if larva present 2.006 11 Eriocrania cicatricella Yes 4 R Easier if larva present 2.007 13 Eriocrania semipurpurella Yes 4 R Easier if larva present 2.008 12 Eriocrania sangii Yes 4 R Easier if larva present 4.001 118 Enteucha acetosae 0 A 4.002 116 Stigmella lapponica 0 L 4.003 117 Stigmella confusella 0 L 4.004 90 Stigmella tiliae 0 A 4.005 110 Stigmella betulicola 0 L 4.006 113 Stigmella sakhalinella 0 L 4.007 112 Stigmella luteella 0 L 4.008 114 Stigmella glutinosae 0 L Examination of larva essential 4.009 115 Stigmella alnetella 0 L Examination of larva essential 4.010 111 Stigmella microtheriella Yes 0 L 4.011 109 Stigmella prunetorum 0 L 4.012 102 Stigmella aceris 0 A 4.013 97 Stigmella malella Apple Pigmy 0 L 4.014 98 Stigmella catharticella 0 A 4.015 92 Stigmella anomalella Rose Leaf Miner 0 L 4.016 94 Stigmella spinosissimae 0 R 4.017 93 Stigmella centifoliella 0 R 4.018 80 Stigmella ulmivora 0 L Exit-hole must be shown or larval colour 4.019 95 Stigmella viscerella -

Morphology, Taxonomy, and Biology of Larval Scarabaeoidea
Digitized by the Internet Archive in 2011 with funding from University of Illinois Urbana-Champaign http://www.archive.org/details/morphologytaxono12haye ' / ILLINOIS BIOLOGICAL MONOGRAPHS Volume XII PUBLISHED BY THE UNIVERSITY OF ILLINOIS *, URBANA, ILLINOIS I EDITORIAL COMMITTEE John Theodore Buchholz Fred Wilbur Tanner Charles Zeleny, Chairman S70.S~ XLL '• / IL cop TABLE OF CONTENTS Nos. Pages 1. Morphological Studies of the Genus Cercospora. By Wilhelm Gerhard Solheim 1 2. Morphology, Taxonomy, and Biology of Larval Scarabaeoidea. By William Patrick Hayes 85 3. Sawflies of the Sub-family Dolerinae of America North of Mexico. By Herbert H. Ross 205 4. A Study of Fresh-water Plankton Communities. By Samuel Eddy 321 LIBRARY OF THE UNIVERSITY OF ILLINOIS ILLINOIS BIOLOGICAL MONOGRAPHS Vol. XII April, 1929 No. 2 Editorial Committee Stephen Alfred Forbes Fred Wilbur Tanner Henry Baldwin Ward Published by the University of Illinois under the auspices of the graduate school Distributed June 18. 1930 MORPHOLOGY, TAXONOMY, AND BIOLOGY OF LARVAL SCARABAEOIDEA WITH FIFTEEN PLATES BY WILLIAM PATRICK HAYES Associate Professor of Entomology in the University of Illinois Contribution No. 137 from the Entomological Laboratories of the University of Illinois . T U .V- TABLE OF CONTENTS 7 Introduction Q Economic importance Historical review 11 Taxonomic literature 12 Biological and ecological literature Materials and methods 1%i Acknowledgments Morphology ]* 1 ' The head and its appendages Antennae. 18 Clypeus and labrum ™ 22 EpipharynxEpipharyru Mandibles. Maxillae 37 Hypopharynx <w Labium 40 Thorax and abdomen 40 Segmentation « 41 Setation Radula 41 42 Legs £ Spiracles 43 Anal orifice 44 Organs of stridulation 47 Postembryonic development and biology of the Scarabaeidae Eggs f*' Oviposition preferences 48 Description and length of egg stage 48 Egg burster and hatching Larval development Molting 50 Postembryonic changes ^4 54 Food habits 58 Relative abundance. -

Butterfly Checklist 08
Family Hesperiwidae (Skippers) Subfamily Hesperiinae (Grass Skippers) Subfamily Pyrginae (Spread-wing Skippers) ____Julia’s Skipper Nastra julia ____ Guava Skipper Phocides polbius ____Pale-rayed Skipper Vidius perigenes ____Mercurial Skipper Proteides mercurius ____Fawn-spotted Skipper Cymaenes odilia ____White-striped Longtail Chioides catillus ____Clouded Skipper Lerema accius ____Zilpa Longtail Chioides zilpa ____Orange Skipperling Copaeodes aurantiacia ____Long-tailed Skipper Urbanus proteus ____Southern Skipperling Copaeodes minima ____Teleus Longtail Urbanus teleus ____Fiery Skipper Hylephila phyleus ) ____Brown Longtail Urbanus procne ____Whirlabout Polites vibex ____ Dorantes Longtail Urbanus dorantes ____Southern Broken-Dash Wallengrenia otho ____Coyote Cloudywing Achalarus toxeus ____Sachem Atalopedes campestris ____Mimosa Skipper Cogia calchas ____Celia's Roadside-Skipper Amblyscirtes celia ____Potrillo Skipper Cabares potrillo ____Nysa Roadside-Skipper Amblyscirtes nyhsa ____Two-barred Flasher Astraptes fulgerator ____Dun Skipper Euphyes vestries ____Glazed Pellicia Pellicia arina ____Eufala Skipper Lerodea eufala ____Olive-clouded Skipper Lerodea arabus ____Mazans Scallopwing Staphylus mazans ____Texas Powdered Skipper Systasea pulverulenta ____Brazilian Skipper Calpodes ethlius ____Sickle-winged Skipper Eantis tamenund ____Obscure Skipper Panoquina panoquinoides ____Brown-banded Skipper Timochares ruptifasciatus ____Ocola Skipper Panoquina ocola ____White-patched Skipper Chiomara georgina ____Purple-washed Skipper Panoquina -

A Simple 2D Non-Parametric Resampling Statistical Approach To
A Simple 2D Non-Parametric Resampling Statistical Approach to Assess Confidence in Species Identification in DNA Barcoding—An Alternative to Likelihood and Bayesian Approaches Qian Jin1, Li-Jun He2, Ai-Bing Zhang1* 1 College of Life Sciences, Capital Normal University, Beijing, P. R. China, 2 State Key Laboratory of Estuarine and Coastal Research, East China Normal University, Shanghai, P. R. China Abstract In the recent worldwide campaign for the global biodiversity inventory via DNA barcoding, a simple and easily used measure of confidence for assigning sequences to species in DNA barcoding has not been established so far, although the likelihood ratio test and the Bayesian approach had been proposed to address this issue from a statistical point of view. The TDR (Two Dimensional non-parametric Resampling) measure newly proposed in this study offers users a simple and easy approach to evaluate the confidence of species membership in DNA barcoding projects. We assessed the validity and robustness of the TDR approach using datasets simulated under coalescent models, and an empirical dataset, and found that TDR measure is very robust in assessing species membership of DNA barcoding. In contrast to the likelihood ratio test and Bayesian approach, the TDR method stands out due to simplicity in both concepts and calculations, with little in the way of restrictive population genetic assumptions. To implement this approach we have developed a computer program package (TDR1.0beta) freely available from ftp://202.204.209.200/education/video/TDR1.0beta.rar. Citation: Jin Q, He L-J, Zhang A-B (2012) A Simple 2D Non-Parametric Resampling Statistical Approach to Assess Confidence in Species Identification in DNA Barcoding—An Alternative to Likelihood and Bayesian Approaches. -

Journal of the Lepidopterists' Society
JOURNAL OF THE LEPIDOPTERISTS' SOCIETY Volume 38 1984 Number 3 Joumal of the Lepidopterists' Society 38(3). 1984. 149-164 SOD WEBWORM MOTHS (PYRALIDAE: CRAMBINAE) IN SOUTH DAKOTA B. McDANIEL,l G. FAUSKEl AND R. D. GUSTIN 2 ABSTRACT. Twenty-seven species of the subfamily Crambinae known as sod web worm moths were collected from South Dakota. A key to species has been included as well as their distribution patterns in South Dakota. This study began after damage to rangeland in several South Dakota counties in the years 1974 and 1975. Damage was reported from Cor son, Dewey, Harding, Haakon, Meade, Perkins, Stanley and Ziebach counties. An effort was made to determine the species of Crambinae present in South Dakota and their distribution. Included are a key for species identification and a list of species with their flight periods and collection sites. MATERIALS AND METHODS Black light traps using the General Electric Fluorescent F ls T8 B1 15 watt bulb were set up in Brookings, Jackson, Lawrence, Minnehaha, Pennington and Spink counties. In Minnehaha County collecting was carried out with a General Electric 200 watt soft-glow bulb. Daytime collecting was used in several localities. Material in the South Dakota State University Collection was also utilized. For each species a map is included showing collection localities by county. On the maps the following symbols are used: • = collected by sweepnet. Q = collected by light trap. Key to South Dakota Cram binae 1a. Rs stalked .__ ... ___ .. __ ......................... _..... _ ................................. _._............................................. 2 lb. Rs arising directly from discal cell ................................................................. _............ _............ -

Annotated Checklist of the Butterflies of Bentsen-Rio Grande Valley State
AN ANNOTATED CHECKLIST OF THE BUTTERFLIES (LEPIDOPTERA: RHOPALOCERA) OF BENTSEN-RIO GRANDE STATE VALLEY PARK AND VICINITY JUNE, 1974 Published by TEXAS PARKS & WILDLIFE DEPARTMENT BENTSEN-RIO GRANDE VALLEY STATE PARK P.O. 30X 988; MISSION, TEXAS 78572 INTRODUCTION The species listed here in are primarily a result of the collecting by the authors during the period 1972-1973. Certain important records of the previous several years are also included. Additionally, the checklist incorporates records of a number of other lepidopterists. The primary focus of the checklist, then, is upon recent collecting, rather than being an attempt to list all known records from the Mid-Valley area. All lepidopterists collecting in the park and vicinity are urged to send copies of their records to the authors and/or the park authorities. A number of species on the list have been taken in Hidalgo Co. but not yet within the actual confines of the park; the annotations will indicate which species these are. Some of these have been taken at Santa Ana National Wildlife Refuge, approximately thirty miles down river, in habitats similar to those within the park. Others have been taken within several miles of the park, in nearby towns and along roadsides. These species can be reasonably expected to occur in the park, and their inclusion upon this list should alert the collector to their possible presence. The annotations have been kept necessarily brief. They are intended to aid the visiting lepidopterist in evaluating the significance of his catches. Local larval food plants are given where known. Much, however, is still to be learned regarding the life histories of even some of the commoner species. -
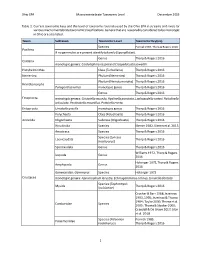
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 1 Table 1. Current Taxonomic Keys and the Level of Taxonomy Routinely U
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Table 1. Current taxonomic keys and the level of taxonomy routinely used by the Ohio EPA in streams and rivers for various macroinvertebrate taxonomic classifications. Genera that are reasonably considered to be monotypic in Ohio are also listed. Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Species Pennak 1989, Thorp & Rogers 2016 Porifera If no gemmules are present identify to family (Spongillidae). Genus Thorp & Rogers 2016 Cnidaria monotypic genera: Cordylophora caspia and Craspedacusta sowerbii Platyhelminthes Class (Turbellaria) Thorp & Rogers 2016 Nemertea Phylum (Nemertea) Thorp & Rogers 2016 Phylum (Nematomorpha) Thorp & Rogers 2016 Nematomorpha Paragordius varius monotypic genus Thorp & Rogers 2016 Genus Thorp & Rogers 2016 Ectoprocta monotypic genera: Cristatella mucedo, Hyalinella punctata, Lophopodella carteri, Paludicella articulata, Pectinatella magnifica, Pottsiella erecta Entoprocta Urnatella gracilis monotypic genus Thorp & Rogers 2016 Polychaeta Class (Polychaeta) Thorp & Rogers 2016 Annelida Oligochaeta Subclass (Oligochaeta) Thorp & Rogers 2016 Hirudinida Species Klemm 1982, Klemm et al. 2015 Anostraca Species Thorp & Rogers 2016 Species (Lynceus Laevicaudata Thorp & Rogers 2016 brachyurus) Spinicaudata Genus Thorp & Rogers 2016 Williams 1972, Thorp & Rogers Isopoda Genus 2016 Holsinger 1972, Thorp & Rogers Amphipoda Genus 2016 Gammaridae: Gammarus Species Holsinger 1972 Crustacea monotypic genera: Apocorophium lacustre, Echinogammarus ischnus, Synurella dentata Species (Taphromysis Mysida Thorp & Rogers 2016 louisianae) Crocker & Barr 1968; Jezerinac 1993, 1995; Jezerinac & Thoma 1984; Taylor 2000; Thoma et al. Cambaridae Species 2005; Thoma & Stocker 2009; Crandall & De Grave 2017; Glon et al. 2018 Species (Palaemon Pennak 1989, Palaemonidae kadiakensis) Thorp & Rogers 2016 1 Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Informal grouping of the Arachnida Hydrachnidia Smith 2001 water mites Genus Morse et al.