A Simple 2D Non-Parametric Resampling Statistical Approach To
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Butterfly Checklist 08
Family Hesperiwidae (Skippers) Subfamily Hesperiinae (Grass Skippers) Subfamily Pyrginae (Spread-wing Skippers) ____Julia’s Skipper Nastra julia ____ Guava Skipper Phocides polbius ____Pale-rayed Skipper Vidius perigenes ____Mercurial Skipper Proteides mercurius ____Fawn-spotted Skipper Cymaenes odilia ____White-striped Longtail Chioides catillus ____Clouded Skipper Lerema accius ____Zilpa Longtail Chioides zilpa ____Orange Skipperling Copaeodes aurantiacia ____Long-tailed Skipper Urbanus proteus ____Southern Skipperling Copaeodes minima ____Teleus Longtail Urbanus teleus ____Fiery Skipper Hylephila phyleus ) ____Brown Longtail Urbanus procne ____Whirlabout Polites vibex ____ Dorantes Longtail Urbanus dorantes ____Southern Broken-Dash Wallengrenia otho ____Coyote Cloudywing Achalarus toxeus ____Sachem Atalopedes campestris ____Mimosa Skipper Cogia calchas ____Celia's Roadside-Skipper Amblyscirtes celia ____Potrillo Skipper Cabares potrillo ____Nysa Roadside-Skipper Amblyscirtes nyhsa ____Two-barred Flasher Astraptes fulgerator ____Dun Skipper Euphyes vestries ____Glazed Pellicia Pellicia arina ____Eufala Skipper Lerodea eufala ____Olive-clouded Skipper Lerodea arabus ____Mazans Scallopwing Staphylus mazans ____Texas Powdered Skipper Systasea pulverulenta ____Brazilian Skipper Calpodes ethlius ____Sickle-winged Skipper Eantis tamenund ____Obscure Skipper Panoquina panoquinoides ____Brown-banded Skipper Timochares ruptifasciatus ____Ocola Skipper Panoquina ocola ____White-patched Skipper Chiomara georgina ____Purple-washed Skipper Panoquina -

Annotated Checklist of the Butterflies of Bentsen-Rio Grande Valley State
AN ANNOTATED CHECKLIST OF THE BUTTERFLIES (LEPIDOPTERA: RHOPALOCERA) OF BENTSEN-RIO GRANDE STATE VALLEY PARK AND VICINITY JUNE, 1974 Published by TEXAS PARKS & WILDLIFE DEPARTMENT BENTSEN-RIO GRANDE VALLEY STATE PARK P.O. 30X 988; MISSION, TEXAS 78572 INTRODUCTION The species listed here in are primarily a result of the collecting by the authors during the period 1972-1973. Certain important records of the previous several years are also included. Additionally, the checklist incorporates records of a number of other lepidopterists. The primary focus of the checklist, then, is upon recent collecting, rather than being an attempt to list all known records from the Mid-Valley area. All lepidopterists collecting in the park and vicinity are urged to send copies of their records to the authors and/or the park authorities. A number of species on the list have been taken in Hidalgo Co. but not yet within the actual confines of the park; the annotations will indicate which species these are. Some of these have been taken at Santa Ana National Wildlife Refuge, approximately thirty miles down river, in habitats similar to those within the park. Others have been taken within several miles of the park, in nearby towns and along roadsides. These species can be reasonably expected to occur in the park, and their inclusion upon this list should alert the collector to their possible presence. The annotations have been kept necessarily brief. They are intended to aid the visiting lepidopterist in evaluating the significance of his catches. Local larval food plants are given where known. Much, however, is still to be learned regarding the life histories of even some of the commoner species. -

Mitochondrial Pseudogenes in Insect DNA Barcoding: Differing Points of View on the Same Issue
Biota Neotrop., vol. 12, no. 3 Mitochondrial pseudogenes in insect DNA barcoding: differing points of view on the same issue Luis Anderson Ribeiro Leite1,2 1Laboratório de Estudos de Lepidoptera Neotropical, Departamento de Zoologia, Universidade Federal do Paraná – UFPR, CP 19020, CEP 81531-980, Curitiba, PR, Brasil 2Corresponding author: Luis Anderson Ribeiro Leite, e-mail: [email protected] LEITE, L.A.R. Mitochondrial pseudogenes in insect DNA barcoding: differing points of view on the same issue. Biota Neotrop. 12(3): http://www.biotaneotropica.org.br/v12n3/en/abstract?thematic-review+bn02412032012 Abstract: Molecular tools have been used in taxonomy for the purpose of identification and classification of living organisms. Among these, a short sequence of the mitochondrial DNA, popularly known as DNA barcoding, has become very popular. However, the usefulness and dependability of DNA barcodes have been recently questioned because mitochondrial pseudogenes, non-functional copies of the mitochondrial DNA incorporated into the nuclear genome, have been found in various taxa. When these paralogous sequences are amplified together with the mitochondrial DNA, they may go unnoticed and end up being analyzed as if they were orthologous sequences. In this contribution the different points of view regarding the implications of mitochondrial pseudogenes for entomology are reviewed and discussed. A discussion of the problem from a historical and conceptual perspective is presented as well as a discussion of strategies to keep these nuclear mtDNA copies out of sequence analyzes. Keywords: COI, molecular, NUMTs. LEITE, L.A.R. Pseudogenes mitocondriais no DNA barcoding em insetos: diferentes pontos de vista sobre a mesma questão. -

In Costa Rica
GENERAL NOTES Journal of the Lepidopterists' Society 39(3), 1985, 215-223 NATURAL HISTORY NOTES ON ASTRAPTES AND URBANUS (HESPERIIDAE) IN COSTA RICA The close evolutionary affinity between Astraptes and Urban us skippers (Hesperiidae) as members of the "Urbanus group" within the Pyrginae (Evans, 1952, Catalogue of the American Hesperiidae. Part II, British Museum of Natural History, London, 178 pp.) suggests similarities in the comparative biology of immature stages among representative species in both genera. For example, published larval food plant records for both genera include Leguminosae, and Astraptes has only been found feeding on members of this family (e.g., Howe, 1975, The butterflies of North America, Doubleday & Co., New York, 591 pp.; Kendall, 1976, Bull. Allyn Mus. No. 39, 9 pp.). While the majority of Urbanus species are legume-feeders as caterpillars, a handful of species are grass-feeders (Howe, op. cit.). Further field studies on the natural history of selected species in both genera may either confirm existing patterns of larval food plant patterns or augment them with records involving yet other families of dicotyledonous plants. It is evident that, within the Hesperiidae, tropical genera and species have undergone considerable evolutionary divergence in terms of larval food plants, with ten or more distinct food plant families known for some regions of the Neotropics (e.g., Kendall, op. cit.). In this note I summarize life cycle and larval food plant notes for A. Julgerator (Walch) in Costa Rica and make some brief comparisons with similar data on U. proteus (Linnaeus) from the same or similar localities within the country. -

Moths and Butterflies
LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004MOTHS LJL©2004 LJL©2004AND BUTTERFLIES LJL©2004 LJL©2004 (LEPIDOPTERA) LJL©2004 LJL©2004 LJL©2004 FROM LJL©2004 BAHÍA LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004HONDA LJL©2004 LJL©2004 AND CANALES LJL©2004 LJL©2004 DE TIERRA LJL©2004 ISLANDLJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004(VERAGUAS, LJL©2004 LJL©2004 PANAMA LJL©2004) LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 LJL©2004 -

The Barcode of Life Initiative: Reply to Dupré, Hollingsworth and Holm
Genomics, Society and Policy 2007, Vol.3, No.2, pp.52-56 The Barcode of Life Initiative: Reply to Dupré, Hollingsworth and Holm FILIPE O. COSTA AND GARY R. CARVALHO1 We are grateful to John Dupré, Peter Hollingsworth and Petter Holm for their insightful and constructive responses to our article.2 As with any new and increasingly applied approach, DNA barcoding has provoked considerable discussion, even though the basic technology employed is essentially a refinement of existing molecular approaches to systematics.3 What characterises DNA barcoding is the attempt to standardise the molecular approach by focusing on one or a few genes with appropriate levels of among-species divergence, and to secure global accessibility to a common database. Additionally, although one gene, cytochrome oxidase I (COI), has proven to be informative across diverse taxa, the aim of DNA barcoding has not been to identify a single common gene, but rather to maximise standardisation across related taxa to ensure high comparability. DNA barcoding is essentially a practical tool that can be applied to compare a target DNA sequence with a reference DNA sequence that may confirm species identity or generate alternative hypotheses of species delineation. It is crucial therefore to appreciate that rather than replacing conventional approaches to taxonomy, which rely heavily on ecological, morphological and behavioural characteristics, DNA barcoding can in many cases render the Linnaean system more accessible. A recent cover of Nature4 illustrating a modern-age Linnaeus wearing a contemporary naturalist’s outfit and holding a barcode in his hand could not be more paradigmatic. Rather than rehearse many previous discussions and articles on the merits and limitations of DNA barcoding, here we focus on just a few of the key points raised by Dupré, Hollingsworth and Holm. -

TPSS-2008-Vol1n2 354Russell.Pdf (7.920Mb)
University of Plymouth PEARL https://pearl.plymouth.ac.uk The Plymouth Student Scientist - Volume 01 - 2008 The Plymouth Student Scientist - Volume 1, No. 2 - 2008 2008 Taking Stock of the World's Species Russell, S. Russell, S. (2008) 'Taking Stock of the World's Species [POSTER]', The Plymouth Student Scientist, 1(2), p. 354. http://hdl.handle.net/10026.1/13821 The Plymouth Student Scientist University of Plymouth All content in PEARL is protected by copyright law. Author manuscripts are made available in accordance with publisher policies. Please cite only the published version using the details provided on the item record or document. In the absence of an open licence (e.g. Creative Commons), permissions for further reuse of content should be sought from the publisher or author. TAKING STOCK OF THE W O R L D ’ S S P E C I E S Wouldn’t it be great if every animal and plant had an easy-to-read label, telling you to which species it belongs? 0 1 6 9 6 2 1 2 4 5 6 8 3 Introduction by Samantha Russell Scientists are to establish a giant catalogue of life - to, in effect, “barcode” every species on Earth. "is poster aims to provide an overview of DNA barcoding: what is it? and why is it controversial? A barcode for biodiversity Promises and pitfalls A barcode is a machine-readable digital tag that can identify items to a Promise: "TJOHMFHFOFJTTFRVFODFEGPSVTFBTUIFCBSDPEF VTFGVMMFWFMPGVOJRVFOFTT<> To be used universally allowing standardization of protocols [3]. ɨFJOUFOUPG%/"CBSDPEJOHJTUPVTFBTIPSU%/"TFRVFODFGSPNB Pitfall: No single gene will work for all taxa. -

DNA Barcode Sequences Are Very Short Relative to the Entire Genome and They Can Be Obtained Reasonably Quickly and Cheaply
DNA barcoding is a technique for characterizing species of organisms using a short DNA sequence from a standard and agreed-upon position in the genome. DNA barcode sequences are very short relative to the entire genome and they can be obtained reasonably quickly and cheaply. The cytochrome c oxidase subunit 1 mitochondrial region (COI) is emerging as the standard barcode region for higher animals. It is 648 nucleotide base pairs long in most groups, a very short sequence relative to 3 billion base pairs in the human genome, for example. The ‘barcode’ metaphor is useful though not correct in fine detail. That is, all the products of one type on a supermarket shelf (like a 2- litre bottle of Coca-Cola) share exactly the same 11-digit barcode, which is distinct from all other barcodes. DNA barcodes vary among individuals of the same species, but only to a very minor degree. If the DNA barcode region is effective, the minor variation within species will be much smaller than the differences among species. DNA barcoding is a taxonomic method that uses a short genetic marker in an organism's DNA to identify it as belonging to a particular species. It differs from molecular phylogeny in that the main goal is not to determine classification but to identify an unknown sample in terms of a known classification.[1] Although barcodes are sometimes used in an effort to identify unknown species or assess whether species should be combined or separated,[2] the utility of DNA barcoding for these purposes is subject to debate.[3] Why barcode animal and plant species? By harnessing advances in electronics and genetics, barcoding will help many people quickly and cheaply recognize known species and retrieve information about them speed discovery of the millions of species yet to be named provide vital new tools for appreciating and managing the Earth’s immense and changing biodiversity. -

Problems with DNA Barcodes for Species
Systematics and Biodiversity 4 (2): 127–132 Issued 7 June 2006 doi:10.1017/S147720000500191X Printed in the United Kingdom C The Natural History Museum Andrew V.Z. Brower PERSPECTIVE Department of Zoology, Oregon State University, Corvallis, OR 97331, USA Email: browera@science. Problems with DNA barcodes for species oregonstate.edu delimitation: ‘ten species’ of Astraptes submitted October 2005 accepted December 2005 fulgerator reassessed (Lepidoptera: Hesperiidae) Abstract Hebert and colleagues (2004) used a short region of the mitochondrial Cytochrome oxidase subunit I gene as a delimiter for ten putative species from among 466 individuals of the skipper butterfly currently known as Astraptes ful- gerator from Guanacaste, Costa Rica. Their data are reanalysed to assess cluster stability and clade support using Neighbor-Joining bootstrap, population aggrega- tion analysis and cladistic haplotype analysis. At least three, but not more than seven mtDNA clades that may correspond to cryptic species are supported by the evidence. Additional difficulties with Hebert et al.’s interpretation of the data are discussed. Key words mtDNA, COI, molecular systematics, skipper butterfly Introduction Using molecular data for species identification has been used in forensics for almost 20 years (Higuchi et al., 1988; For some years now, the limiting stage in study of DNA data Li et al., 1988; Sperling et al., 1994; Baker & Palumbi, 1994; has not been the generation of sequences themselves, which DeSalle & Birstein, 1996), and has been employed for identi- is now routinely performed at an industrial scale, or even out- fying various closely related insects and associating holometa- sourced to private companies, much like sending a roll of bolous life-stages by a number of authors (Sperling et al. -
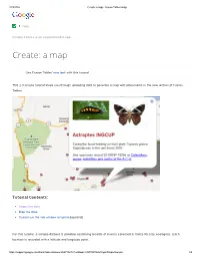
Ate: a Map - Fusion Tables Help
2/19/2014 Create: a map - Fusion Tables Help Help Community Fusion Tables is an experimental app. Create: a map Use Fusion Tables' new look with this tutorial This 2-3 minute tutorial steps you through uploading data to generate a map with placemarks in the new version of Fusion Tables. Tutorial Contents: Import the data Map the data Customize the info window template [optional] For this tutorial, a sample dataset is provided containing records of insects collected in Costa Rica by ecologists. Each location is recorded with a latitude and longitude point. https://support.google.com/fusiontables/answer/2527132?hl=en&topic=2573107&ctx=topic#importsample 1/5 2/19/2014 Create: a map - Fusion Tables Help Import the sample dataset 1. Download this sample insect dataset. 2. Go to Google Docs. Sign in to your Google Account or create a Google Account if you don't already have one. (Note that you can't use a Google Apps for your Domain account for Fusion Tables.) 3. Click the Create button. 4. Choose Create > More > Fusion Table from the dropdown menu. 5. In the Import new table dialog box, click Choose File. 6. Select the "Astraptes fulgerator complex sample data.csv" file you downloaded, and press Next. 7. Check that the data is formatted correctly and click Next. 8. Give your table a name and click Finish. Your uploaded data now appears in a new Fusion Table with thumbnail images in the two columns of URL links: Map the data Fusion Tables auto-detects location data in a table and displays a tab called "Map of <location column name>." In this case, the Map tab is titled "Map of latitude." 1. -

Morphological and Molecular Analysis of Australian Earwigs (Dermaptera) Points to Unique Species and Regional Endemism in the Anisolabididae Family
insects Article Morphological and Molecular Analysis of Australian Earwigs (Dermaptera) Points to Unique Species and Regional Endemism in the Anisolabididae Family Oliver P. Stuart 1 , Matthew Binns 1,2, Paul A. Umina 1,3, Joanne Holloway 4 , Dustin Severtson 5, Michael Nash 6,7 , Thomas Heddle 8 , Maarten van Helden 6,8 and Ary A. Hoffmann 1,* 1 School of BioSciences, Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Parkville, Victoria 3052, Australia; [email protected] (O.P.S.); [email protected] (M.B.); [email protected] (P.A.U.) 2 Agriculture and Food Business Unit, Commonwealth Scientific and Industrial Research Organisation, Black Mountain, Australian Capital Territory 2601, Australia 3 Cesar, 293 Royal Parade, Parkville, Victoria 3052, Australia 4 New South Wales Department of Primary Industries, Wagga Wagga Agricultural Institute, Pine Gully Road, Charles Sturt University, New South Wales 2795, Australia; [email protected] 5 Department of Primary Industries and Regional Development, South Perth, Western Australia 6151, Australia; [email protected] 6 School of Agriculture, Food and Wine, the University of Adelaide, Urrbrae, South Australia 5064, Australia; [email protected] (M.N.); [email protected] (M.v.H.) 7 School of Life Science, College of Science, Health and Engineering, La Trobe University, Bundoora, Victoria 3086, Australia 8 South Australian Research and Development Institute, Entomology, Waite Road, Waite, Urrbrae, South Australia 5064, Australia; [email protected] * Correspondence: [email protected] Received: 1 February 2019; Accepted: 7 March 2019; Published: 14 March 2019 Abstract: Dermaptera (earwigs) from the Anisolabididae family may be important for pest control but their taxonomy and status in Australia is poorly studied. -

DNA Barcode Authentication of Wood Samples of Threatened and Commercial Timber Trees Within the Tropical Dry Evergreen Forest of India
DNA Barcode Authentication of Wood Samples of Threatened and Commercial Timber Trees within the Tropical Dry Evergreen Forest of India Stalin Nithaniyal1,2, Steven G. Newmaster3*, Subramanyam Ragupathy3, Devanathan Krishnamoorthy1, Sophie Lorraine Vassou1, Madasamy Parani1* 1 Department of Genetic Engineering, Center for DNA Barcoding, SRM University, Chennai, India, 2 Interdisciplinary School of Indian System of Medicine, SRM University, Chennai, India, 3 Centre for Biodiversity Genomics, University of Guelph, Guelph, Ontario, Canada Abstract Background: India is rich with biodiversity, which includes a large number of endemic, rare and threatened plant species. Previous studies have used DNA barcoding to inventory species for applications in biodiversity monitoring, conservation impact assessment, monitoring of illegal trading, authentication of traded medicinal plants etc. This is the first tropical dry evergreen forest (TDEF) barcode study in the World and the first attempt to assemble a reference barcode library for the trees of India as part of a larger project initiated by this research group. Methodology/Principal Findings: We sampled 429 trees representing 143 tropical dry evergreen forest (TDEF) species, which included 16 threatened species. DNA barcoding was completed using rbcL and matK markers. The tiered approach (1st tier rbcL;2nd tier matK) correctly identified 136 out of 143 species (95%). This high level of species resolution was largely due to the fact that the tree species were taxonomically diverse in the TDEF. Ability to resolve taxonomically diverse tree species of TDEF was comparable among the best match method, the phylogenetic method, and the characteristic attribute organization system method. Conclusions: We demonstrated the utility of the TDEF reference barcode library to authenticate wood samples from timber operations in the TDEF.