Activating Kinase Mutations in Melanoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bird Trip Report, 11-12 March 2017 BIRDING in BRASSO SECO by Matt Kelly
Quarterly Bulletin of the Trinidad and Tobago Field Naturalists’ Club April – June 2017 Issue No: 2/2017 Bird Trip Report, 11-12 March 2017 BIRDING IN BRASSO SECO by Matt Kelly Selwyn Gomes and I left Port of Spain Community Museum in Arima, which was very at 7:30am on Saturday, March 11, 2017 and interesting. To do justice, an entire article needs to headed over the Northern Range for Brasso be devoted to that community. Seco. We meandered a bit along the way, first stopping at the Santa Rosa First Peoples’ (Continued on page 3) Male tufted coquette Photo by Matt Kelly Page 2 THE FIELD NATURALIST Issue No. 2/2017 Inside This Issue BIRD TRIP REPORT 1 BRASSO SECO Quarterly Bulletin of the - Matt Kelly Trinidad and Tobago Field Naturalists’ Club 50 YEARS OF AWNC April - June 2017 5 - Johanne Ryan Editors 8 IF YOU’VE GOT, IT FLAUNT IT Amy Deacon, Renoir Auguste, -Christopher K. Starr Associate Editor: Rupert Mends ‘NATURALIST IN’ SERIES Contributing writers BETWEEN ATLANTIC TIDES 9 Jessica Rozek, Christopher K. Starr, Hans Boos, Kris Sookdeo, - Christopher K Starr Renoir Auguste, Matt Kelly, Feroze Omardeen, Reynold Boyce, Johanne Ryan BIRD GROUP REPORT 11 GRAND COUVA Photographs - Matt Kelly Jessica Rozek, Aly Olliviere, Jeffrey Wong Sang, 13 YOUR IDEAS AND OBSERVATIONS Hans Boos, Kris Sookdeo, Christopher K. Starr, Renoir Auguste, Matt Kelly, Faraaz Abdool, Feroze Omardeen, Asif FIELD TRIP REPORT Khan, Kenneth Fournillier, Harold Diaz. 14 COCOA ESTATE AT FISHING POND - Reynold Boyce Design and Layout 16 CONFERENCE REPORT Eddison Baptiste and Amy Deacon CELEBRATING CARIBBEAN BIRD DIVERSITY - Jessica Rozek 18 STRATEGIC PLAN UPDATE - Compiled by Amy Deacon 19 ‘YOU SHOULDN’T BE IN SCHOOL’ The Trinidad and Tobago Field Naturalists’ Club Presentation of Elisha Tikasingh’s Memoirs is a non-profit, non-governmental organization 20 NATURE IN THE NEWS Management Committee 2017/2018 Compiled by Kris Sookdeo President …………….. -

United Nations List of Delegations to the Second High-Level United
United Nations A/CONF.235/INF/2 Distr.: General 30 August 2019 Original: English Second High-level United Nations Conference on South-South Cooperation Buenos Aires, 20–22 March 2019 List of delegations to the second High-level United Nations Conference on South-South Cooperation 19-14881 (E) 110919 *1914881* A/CONF.235/INF/2 I. States ALBANIA H.E. Mr. Gent Cakaj, Acting Minister for Europe and Foreign Affairs H.E. Ms. Besiana Kadare, Ambassador, Permanent Representative Mr. Dastid Koreshi, Chief of Staff of the Acting Foreign Minister ALGERIA H.E. Mr. Abdallah Baali, Ambassador Counsellor, Ministry of Foreign Affairs Alternate Head of Delegation H.E. Mr. Benaouda Hamel, Ambassador of Algeria in Argentina, Embassy of Algeria in Argentina Representatives Mr. Nacim Gaouaoui, Deputy Director, Ministry of Foreign Affairs Mr. Zoubir Benarbia, First Secretary, Permanent Mission of Algeria to the United Nations Mr. Mohamed Djalel Eddine Benabdoun, First Secretary, Embassy of Algeria in Argentina ANDORRA Mrs. Gemma Cano Berne, Director for Multilateral Affairs and Cooperation Mrs. Julia Stokes Sada, Desk Officer for International Cooperation for Development ANGOLA H.E. Mr. Manuel Nunes Junior, Minister of State for Social and Economic Development, Angola Representatives H.E. Mr. Domingos Custodio Vieira Lopes, Secretary of State for International Cooperation and Angolan Communities, Angola H.E. Ms. Maria de Jesus dos Reis Ferreira, Ambassador Extraordinary and Plenipotentiary, Permanent Representative, Permanent Mission of Angola to the United Nations ANTIGUA AND BARBUDA H.E. Mr. Walton Alfonso Webson, Ambassador Extraordinary and Plenipotentiary, Permanent Representative, Permanent Mission Representative Mr. Claxton Jessie Curtis Duberry, Third Secretary, Permanent Mission 2/42 19-14881 A/CONF.235/INF/2 ARGENTINA H.E. -

Zoos' New Efforts Put Boots on the Ground to Help Preserve and Protect
BAT CONSERVATION INTERNATIONAL ISSUE 2 • 2016 BATCON.ORG Zoos’ new efforts put boots on the ground to help preserve and protect bats outside their gates BEYONDTHEMENAGERIE SPECIES SPOTLIGHT: WNS RESEARCH BCI WELCOMES THE RODRIGUES FRUIT BAT UPDATE DR. FRICK BECOME a MONTHLY SUSTAINING MEMBER When you set up monthly donations, you allow us to plan our conservation and education programs with confidence, knowing the resources you and other sustaining members provide will be there. Being a Sustaining Member is also convenient for you—your monthly gift is automatically transferred from your debit or credit card or bank account and can be changed or cancelled at any time. It’s safe and secure, and you’re in complete control. You also won’t receive annual membership renewal requests, which help us cut back on paper and postage costs. BCI Sustaining Members receive our Bats magazine, email updates on bat conservation and an opportunity to visit Bracken Cave with up to three guests every year. Your steady support throughout the year allows us to give a voice for bat conservation and helps strengthen our focus on real results. Photos: Jose Martinez Jose Photos: To become a Sustaining Member today visit BATCON.ORG/SUSTAINING or select Sustaining Member on the donation envelope enclosed with your desired monthly gift amount. ISSUE 2 • 2016 bats INSIDE THIS ISSUE FEATURES 08 BEYOND THE MENAGERIE Zoos’ new efforts preserve and protect bats outside their gates Photo: Frank Ridgley OFF THE BAT BAT CHATS BCI Executive Director Andrew First-ever Verne -

Notes and Addresses in Front of Diary
LINLEY SAMBOURNE'S DIARY 1896 Notes and addresses in front of diary: Store Ticket 94657 Cape, Springhurst, Bickley, Kent. Academy Waiters. Moody, Golden. Chalk, 46 Archel Road, West Kensington. Cornwallis, 20 St Oswald's Road, West Brompton. Coals. L & N.W Syndicate, 204 York Rd, N. Boehms Ball at Town Hall Kensington where I met Spearman was Tuesday May 17 1881. The John Griffith Cycle Corporation Oriel House, High St, Coventry 61-63 High Rd, Knightsbridge 118 Newgate St, E.C. 20th Centy Lamp 15/6 nicle, 18/6, 21. aluminium. Apollinaris Company, 4 Stratford Place Oxford St, W. John Alexander Anderson Esq, 11 Abchurch Lane London, City E.C. Guthrie's doctor: Stanley Boyle Esq, M.R.C.S, Harley St W. Green, A.E. 3 Queen's Buildings, High St, Chel't. London address: 3 Wellington Square, S.W. 19 Oct '96, 26 Wellington Square S.W. Fletcher, A. 60 Frederick St, Grays Inn Rd. Grego, 23 Granville Square. Hitchings H.J. Green F. Mrs Taylor, 9 Redbourne St, Tedworth Sq, S.W. Mabel Grant, 5 Duke St, Langham Place. Guthrie, c/o Edward Wormald, Kilkiwan House, Maybole, Ayreshire. Mrs Stephenson's addresses for models; Miss Annie Price, 2 Goodson Road, Fulham. Louise Chalk, 46 Archel Road, West Kensington Maud Easton: above has her address. Miss Price, 21, knows several models. King, 16 Chelverton Road, Putney S.W. Jones Jenny. Brown ? 319 Sauchihall St, Glasgow. Hector Maclean. Photographer. (illeg). Flashlight & book. Crathiewick Lodge, Birdhurst Road S Croydon. Angus D Macdonald, model. 5 Duke St, Langham Place. Emmeline Lofthouse, 5 Park Road, Forest Hill, S.E. -
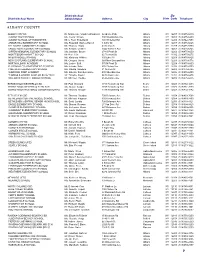
School, Administrator and Address Listing
District/School Zip District/School Name Administrator Address City State Code Telephone ALBANY COUNTY ALBANY CITY SD Dr. Marguerite Vanden Wyngaard Academy Park Albany NY 12207 (518)475-6010 ALBANY HIGH SCHOOL Ms. Cecily Wilson 700 Washington Ave Albany NY 12203 (518)475-6200 ALBANY SCHOOL OF HUMANITIES Mr. C Fred Engelhardt 108 Whitehall Rd Albany NY 12209 (518)462-7258 ARBOR HILL ELEMENTARY SCHOOL Ms. Rosalind Gaines-Harrell 1 Arbor Dr Albany NY 12207 (518)475-6625 DELAWARE COMMUNITY SCHOOL Mr. Thomas Giglio 43 Bertha St Albany NY 12209 (518)475-6750 EAGLE POINT ELEMENTARY SCHOOL Ms. Kendra Chaires 1044 Western Ave Albany NY 12203 (518)475-6825 GIFFEN MEMORIAL ELEMENTARY SCHOOL Ms. Jasmine Brown 274 S Pearl St Albany NY 12202 (518)475-6650 MONTESSORI MAGNET SCHOOL Mr. Ken Lein 65 Tremont St Albany NY 12206 (518)475-6675 MYERS MIDDLE SCHOOL Ms. Kimberly Wilkins 100 Elbel Ct Albany NY 12209 (518)475-6425 NEW SCOTLAND ELEMENTARY SCHOOL Mr. Gregory Jones 369 New Scotland Ave Albany NY 12208 (518)475-6775 NORTH ALBANY ACADEMY Ms. Lesley Buff 570 N Pearl St Albany NY 12204 (518)475-6800 P J SCHUYLER ACHIEVEMENT ACADEMY Ms. Jalinda Soto 676 Clinton Ave Albany NY 12206 (518)475-6700 PINE HILLS ELEMENTARY SCHOOL Ms. Vibetta Sanders 41 N Allen St Albany NY 12203 (518)475-6725 SHERIDAN PREP ACADEMY Ms. Zuleika Sanchez-Gayle 400 Sheridan Ave Albany NY 12206 (518)475-6850 THOMAS S O'BRIEN ACAD OF SCI & TECH Mr. Timothy Fowler 94 Delaware Ave Albany NY 12202 (518)475-6875 WILLIAM S HACKETT MIDDLE SCHOOL Mr. -

Simplified Signs
B Simplifed Signs onvillian Volume 1: Principles, A Manual Sign-Communication System for Special Populations Background, and Application , K Volume 1: Principles, Background, and Application issane John D. Bonvillian, nicole Kissane lee, Tracy T. Dooley, anD Filip T. loncKe l Simplifed Signs presents a system of manual sign communicaton intended for ee Simplifed Signs special populatons who have had limited success mastering spoken or full sign , D languages. It is the culminaton of over twenty years of research and development ooley by the authors. The Simplifed Sign System has been developed and tested for ease of sign comprehension, memorizaton, and formaton by limitng the complexity , of the motor skills required to form each sign, and by ensuring that each sign an visually resembles the meaning it conveys. D l Volume 1 outlines the research underpinning and informing the project, and onc places the Simplifed Sign System in a wider context of sign usage, historically K and by diferent populatons. Volume 2 presents the lexicon of signs, totalling e A Manual Sign-Communication approximately 1000 signs, each with a clear illustraton and a writen descripton of how the sign is formed, as well as a memory aid that connects the sign visually System for Special Populations to the meaning that it conveys. While the Simplifed Sign System originally was developed to meet the needs of s impli persons with intellectual disabilites, cerebral palsy, autsm, or aphasia, it may also assist the communicaton needs of a wider audience – such as healthcare F professionals, aid workers, military personnel , travellers or parents, and children ie D who have not yet mastered spoken language. -

Uv Radiation and Life
European Cooperation in Science and Technology …ESSEM ... UV RADIATION AND LIFE Authors: Zenobia Lity ńska, Alois W. Schmalwieser, Alkiviadis Bais, Karel Ettler, Julian Gröbner, Peter Köpke, Janusz Krzy ścin, Peter den Outer, Jean Verdebout, Gaetano Zipoli and Julita Biszczuk-Jakubowska COST Action 726 Table of content 1. Introduction..................................................................................................... 3 2. Solar UV radiation .......................................................................................... 5 2.1. Factors influencing UV radiation ............................................................... 5 2.2. UV Measurements .................................................................................. 10 2.3. UV modelling........................................................................................... 12 2.4. Geographical distribution and temporal variability of UV radiation in Europe............................................................................................................ 15 3. Biologically effective radiation ................................................................... 21 3.1. Biological effects of UV radiation on human body .................................. 22 3.2. UV Radiation and Animals ...................................................................... 27 3.3. Micro-organisms and UV radiation.......................................................... 30 3.4. UV radiation and Plants ......................................................................... -

Practical Fishermens Congress Comprising The
T O O FFICIA L PUBL ICA I NS . The fo llowing H andb oo k s upon subjects cognate to the u International Fisheri es Ex hib itio n are now p blis h e d . fi or boun d i n clot/i 2s. eat /x . 1 i n llu strat d Wra er Is. eae Demy 8 m, I e pp I H E B FR EDER IC K O LLO Ba rist - at TH E F S R Y L A WS . y P CK , r er C hristi Pro essor of u r L aw M . A . H on. Edi . C or us s , LL D n ; p f J i pruden ce ih the University of O xfo rd. B G EO E B. H O WES Z O O L O G Y A ND FO O D FIS H ES . y RG Demo strator o f o o o rma S cho o o f S c e ce and R o al S cho o o f M es n Bi l gy , N l l i n , y l in outh s n t S Ken i g on . TER FI H . i.ITIS H M A R INE A ND FR ES H WA S ES . Illustr at W A V A utho r o f O f c a G u de . ( ed) By . S ILLE KENT, F fi i l i oo s to th r hto M a chester and Westm ster A uari a . b k e B ig n , n , in q W H H DS W A U B E. -

Strikes on IS in Syria
SUBSCRIPTION SATURDAY, JULY 25, 2015 SHAWWAL 9, 1436 AH No: 16589 Councilor ‘Drifter’ kills 2, Ronaldo dazzles shocked by himself in US as Real rip cleaners’4 plight cinema rampage7 Man48 City apart Turkey stages first air strikes on IS in Syria Min 32º 150 Fils Max 45º Operations follow deal with US on airbase BEYLERBEYI, Turkey: Turkey yesterday vowed to press on with opera- tions against Islamic State (IS) in Syria and other militant groups, after its war planes bombed the jihadists’ positions for the first time. Following the predawn air raids on the IS targets in Syria, Turkish police arrested almost 300 suspected members of IS and pro-Kurdish militant groups nationwide, in one of Turkey’s biggest recent crackdowns on extremists. Prime Minister Ahmet Davutoglu said that the bombing operation had “100 percent” achieved its goals and had “successfully eliminated the targets” targeted by three Turkish F-16s. The air raids were ordered in the wake of a suicide bombing blamed on IS that killed 32 activists on the Syrian border on Monday and cross-border clashes Thursday that claimed the life of a Turkish soldier. The Turkish war planes dropped their charges while in Turkish air- space and did not enter Syrian airspace, the CNN-Turk television channel reported. The Britain-based Syrian Observatory for Human Rights moni- toring group said nine IS fighters were killed in the attacks and 12 wounded. “Turkey will show the strongest reaction to the slightest movement that threatens it,” Davutoglu said. “The operation against IS reached its target and will not stop,” he added. -

RED HACKLE Their Future Starts Here
No. 113 May 2013 THE RED HACKLE their future starts here Boarding Boys & Girls aged 9 to 18 Forces Discount & Bursaries available For more information please contact: Felicity Legge T: 01738 812546 E: [email protected] www.strathallan.co.uk Forgandenny Perthshire PH2 9EG Strathallan is a Scottish Charity dedicated to education. Charity number SC008903 No. 113 42nd 73rd May 2013 THE RED HACKLE The Chronicle of The Black Watch (Royal Highland Regiment), its successor The Black Watch, 3rd Battalion The Royal Regiment of Scotland, The Affiliated Regiments and The Black Watch Association The clay model of the statue that is to stand at Black Watch Corner in Belgium was unveiled in Edinburgh on 3 April 2013. It will commemorate the over 8,000 officers and soldiers of The Black Watch who were killed and over 20,000 who were wounded during the Great War. The artist, Mr Alan Herriot is pictured with the Chairman and some of the Association members. MAY 2013 THE RED HACKLE 1 “I’m stuck at the bridge, I might be late” If you cannot live without reporting your every thought on Facebook and Twitter, then The HALO Trust is not for you. But if you are up for a challenge, want a job with real purpose, then you might find HALO’s charitable work very interesting. HALO’s 8,000 staff clear landmines and unexploded ordnance, and cut up redundant Kalashnikovs, in places in and out of the news. Our international staff are usually in their late 20s or 30s and lead between 250 and 1,000 local employees. -

FEZANA Journal Do Not Necessarily Reflect the Feroza Fitch Views of FEZANA Or Members of This Publication's Editorial Board
FEZANA TABESTAN 1377 AY 3745 ZRE VOL. 22, NO. 2 SUMMER/JUNE 2008 MahJOURJO Amordad Shehrever 1377 AY (Fasli) G Mah Behram-Asfandarmad 1377 AY Farvardin1378 (Shenshai) N G Mah Asfandarmad 1377AL AY Farvardin-Ardehbest 1378 AY (Kadmi) LIVING WITH CHALLENGES Coordinating between 2 Continents & A Special Thank You to Ushta Confectioner's Tandarosti Team Also Inside: Erv. Brgd. Panthaki Invited to Papal Mass Jamsheed Choksy at the White House Farita−Best Female Solo Artist, Australia A Rejuvenated Cama Athornan Institute PUBLICATION OF THE FEDERATION OF ZOROASTRIAN ASSOCIATIONS OF NORTH AMERICA PUBLICATION OF THE FEDERATION OF ZOROASTRIAN ASSOCIATIONS OF NORTH AMERICA Vol 22 No 2 Summer/June 2008 , Tabestan 1377 AY 3746 ZRE President Rustom Kevala, Ph.D. www.fezana.org Editor in Chief: Dolly Dastoor 2 Editorial [email protected] Technical Assistan: Coomi Gazdar Dolly Dastoor Consultant Editor: Lylah M. Alphonse, Message from the [email protected] President Graphic & Layout: Shahrokh Khanizadeh, 4 Financial Report www.khanizadeh.info Cover design: Feroza Fitch, 6 FEZANA Update [email protected] Columnists: Hoshang Shroff:: [email protected] Homi Davier:: [email protected] 13 Living with Challenges Shazneen Rabadi Gandhi : [email protected] 50 Naurooz Around the World Fereshteh Khatibi:: [email protected] Behram Panthaki::[email protected] 73 Women in Finance Behram Pastakia: [email protected] Mahrukh Motafram: [email protected] 76 In The News Nikan Khatibi: [email protected] Copy editors: R Mehta, V Canteenwalla -

2019 Abstracts
11/13/2020 Abstracts | The University of New Orleans 2019 ABSTRACTS Navigate by Department/Specialty* *Note: Visual and performance arts presenters may major in a different discipline. Visit their session pages to review all artistic presentations and abstracts together. Accounting Anthropology & Sociology Biological Science Chemistry Civil & Environmental Engineering Computer Science Earth & Environmental Science Economics & Finance Electrical Engineering English & Foreign Languages History & Philosophy Interdisciplinary Studies Management & Marketing Mathematics Mechanical Engineering Physics Planning & Urban Studies Political Science Psychology Public Administration School of Education School of Hotel, Restaurant, & Tourism School of Naval Architecture & Marine Engineering School of the Arts: Film School of the Arts: Fine Arts School of the Arts: Music School of the Arts: Theatre Accounting E-Central Assessor Ms. Dahlia Daniel Mentor: Cherie Tru mbach November 20 at 10:00-11:30 AM Learning Commons, 1st Floor, #1 02 This product will periodically collect data from each municipal assessor's office, organize, filter and verify the data and then populate an internal proprietary database through API integration or web crawlers. Here, the database will hold the Master Files and be the centralized collection of data from all sources. This data will then be analyzed and reported in a standardized format compatible with an internally managed Geographic Information System (GIS). This allows mapping of attribute data onto geographic layers through a user portal (centralized portal). Customers with a username and password can access the information through mapping and search query functionality. The database will be housed and accessible through third-party server networks, such as Amazon Web Services. The software will be created by a team of developers who will use the Agile methodology to create sprints.