Classification of the Glyphosate Target Enzyme (5-Enolpyruvylshikimate-3-Phosphate Synthase) Lyydia Leino 1,&, Tuoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gut Microbiota and Inflammation
Nutrients 2011, 3, 637-682; doi:10.3390/nu3060637 OPEN ACCESS nutrients ISSN 2072-6643 www.mdpi.com/journal/nutrients Review Gut Microbiota and Inflammation Asa Hakansson and Goran Molin * Food Hygiene, Division of Applied Nutrition, Department of Food Technology, Engineering and Nutrition, Lund University, PO Box 124, SE-22100 Lund, Sweden; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +46-46-222-8327; Fax: +46-46-222-4532. Received: 15 April 2011; in revised form: 19 May 2011 / Accepted: 24 May 2011 / Published: 3 June 2011 Abstract: Systemic and local inflammation in relation to the resident microbiota of the human gastro-intestinal (GI) tract and administration of probiotics are the main themes of the present review. The dominating taxa of the human GI tract and their potential for aggravating or suppressing inflammation are described. The review focuses on human trials with probiotics and does not include in vitro studies and animal experimental models. The applications of probiotics considered are systemic immune-modulation, the metabolic syndrome, liver injury, inflammatory bowel disease, colorectal cancer and radiation-induced enteritis. When the major genomic differences between different types of probiotics are taken into account, it is to be expected that the human body can respond differently to the different species and strains of probiotics. This fact is often neglected in discussions of the outcome of clinical trials with probiotics. Keywords: probiotics; inflammation; gut microbiota 1. Inflammation Inflammation is a defence reaction of the body against injury. The word inflammation originates from the Latin word ―inflammatio‖ which means fire, and traditionally inflammation is characterised by redness, swelling, pain, heat and impaired body functions. -

A Metagenomic Β-Glucuronidase Uncovers a Core Adaptive Function of the Human Intestinal Microbiome
A metagenomic β-glucuronidase uncovers a core adaptive function of the human intestinal microbiome Karine Gloux, Olivier Berteau, Hanane El oumami, Fabienne Béguet, Marion Leclerc, and Joël Doré1 Institut National de la Recherche Agronomique, Unité Mixte de Recherche 1319 Micalis, F-78352 Jouy en Josas, France Edited by Todd R. Klaenhammer, North Carolina State University, Raleigh, NC, and approved June 1, 2010 (received for review February 4, 2010) In the human gastrointestinal tract, bacterial β-D-glucuronidases (BG; XIVa), major reservoirs of BG-positive bacteria (11), are dys- E.C. 3.2.1.31) are involved both in xenobiotic metabolism and in some biosis markers in Crohn’s disease (12–15) and in intestinal carci- of the beneficial effects of dietary compounds. Despite their biolog- nogenesis (16). Nevertheless, the relative contribution of bacterial ical significance, investigations are hampered by the fact that only strains to the global intestinal BG activity remains unclear, and a few BGs have so far been studied. A functional metagenomic the molecular bases are essentially unknown. approach was therefore performed on intestinal metagenomic In the human digestive tract, bacterial BGs are known to be libraries using chromogenic glucuronides as probes. Using this strat- distributed among the Enterobacteriaceae family in some Firmi- egy, 19 positive metagenomic clones were identified but only one cutes genera (Lactobacillus, Streptococcus, Clostridium, Rumino- exhibited strong β-D-glucuronidase activity when subcloned into an coccus, Roseburia, and Faecalibacterium) and in a specific species expression vector. The cloned gene encoded a β-D-glucuronidase of Actinobacteria (Bifidobacterium dentium) (11, 17, 18). On (called H11G11-BG) that had distant amino acid sequence homolo- the basis of their sequence, BGs are highly homologous to gies and an additional C terminus domain compared with known β-galactosidases and only a few of them have been investigated β fi -D-glucuronidases. -
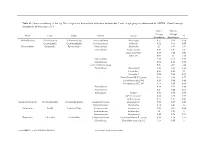
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58 -

Fecal Microbiota Alterations and Small Intestinal Bacterial Overgrowth in Functional Abdominal Bloating/Distention
J Neurogastroenterol Motil, Vol. 26 No. 4 October, 2020 pISSN: 2093-0879 eISSN: 2093-0887 https://doi.org/10.5056/jnm20080 JNM Journal of Neurogastroenterology and Motility Original Article Fecal Microbiota Alterations and Small Intestinal Bacterial Overgrowth in Functional Abdominal Bloating/Distention Choong-Kyun Noh and Kwang Jae Lee* Department of Gastroenterology, Ajou University School of Medicine, Suwon, Gyeonggi-do, Korea Background/Aims The pathophysiology of functional abdominal bloating and distention (FABD) is unclear yet. Our aim is to compare the diversity and composition of fecal microbiota in patients with FABD and healthy individuals, and to evaluate the relationship between small intestinal bacterial overgrowth (SIBO) and dysbiosis. Methods The microbiota of fecal samples was analyzed from 33 subjects, including 12 healthy controls and 21 patients with FABD diagnosed by the Rome IV criteria. FABD patients underwent a hydrogen breath test. Fecal microbiota composition was determined by 16S ribosomal RNA amplification and sequencing. Results Overall fecal microbiota composition of the FABD group differed from that of the control group. Microbial diversity was significantly lower in the FABD group than in the control group. Significantly higher proportion of Proteobacteria and significantly lower proportion of Actinobacteria were observed in FABD patients, compared with healthy controls. Compared with healthy controls, significantly higher proportion of Faecalibacterium in FABD patients and significantly higher proportion of Prevotella and Faecalibacterium in SIBO (+) patients with FABD were found. Faecalibacterium prausnitzii, was significantly more abundant, but Bacteroides uniformis and Bifidobacterium adolescentis were significantly less abundant in patients with FABD, compared with healthy controls. Significantly more abundant Prevotella copri and F. -

Germinants and Their Receptors in Clostridia
JB Accepted Manuscript Posted Online 18 July 2016 J. Bacteriol. doi:10.1128/JB.00405-16 Copyright © 2016, American Society for Microbiology. All Rights Reserved. 1 Germinants and their receptors in clostridia 2 Disha Bhattacharjee*, Kathleen N. McAllister* and Joseph A. Sorg1 3 4 Downloaded from 5 Department of Biology, Texas A&M University, College Station, TX 77843 6 7 Running Title: Germination in Clostridia http://jb.asm.org/ 8 9 *These authors contributed equally to this work 10 1Corresponding Author on September 12, 2018 by guest 11 ph: 979-845-6299 12 email: [email protected] 13 14 Abstract 15 Many anaerobic, spore-forming clostridial species are pathogenic and some are industrially 16 useful. Though many are strict anaerobes, the bacteria persist in aerobic and growth-limiting 17 conditions as multilayered, metabolically dormant spores. For many pathogens, the spore-form is Downloaded from 18 what most commonly transmits the organism between hosts. After the spores are introduced into 19 the host, certain proteins (germinant receptors) recognize specific signals (germinants), inducing 20 spores to germinate and subsequently outgrow into metabolically active cells. Upon germination 21 of the spore into the metabolically-active vegetative form, the resulting bacteria can colonize the 22 host and cause disease due to the secretion of toxins from the cell. Spores are resistant to many http://jb.asm.org/ 23 environmental stressors, which make them challenging to remove from clinical environments. 24 Identifying the conditions and the mechanisms of germination in toxin-producing species could 25 help develop affordable remedies for some infections by inhibiting germination of the spore on September 12, 2018 by guest 26 form. -

Bacteriophages As Potential New Mammalian Pathogens George V
www.nature.com/scientificreports OPEN Bacteriophages as potential new mammalian pathogens George V. Tetz 1, Kelly V. Ruggles2,3, Hua Zhou3, Adriana Heguy4,5,6, Aristotelis Tsirigos3,4,5 & Victor Tetz1 Received: 4 May 2017 Increased intestinal permeability and translocation of gut bacteria trigger various polyaetiological Accepted: 23 June 2017 diseases associated with chronic infammation and underlie a variety of poorly treatable pathologies. Published online: 1 August 2017 Previous studies have established a primary role of the microbiota composition and intestinal permeability in such pathologies. Using a rat model, we examined the efects of exposure to a bacteriophage cocktail on intestinal permeability and relative abundance of taxonomic units in the gut bacterial community. There was an increase in markers of impaired gut permeability, such as the lactulose/mannitol ratio, plasma endotoxin concentrations, and serum levels of infammation- related cytokines, following the bacteriophage challenge. We observed signifcant diferences in the alpha diversity of faecal bacterial species and found that richness and diversity index values increased following the bacteriophage challenge. There was a reduction in the abundance of Blautia, Catenibacterium, Lactobacillus, and Faecalibacterium species and an increase in Butyrivibrio, Oscillospira and Ruminococcus after bacteriophage administration. These fndings provide novel insights into the role of bacteriophages as potentially pathogenic for mammals and their possible implication in the development of diseases associated with increased intestinal permeability. Our knowledge of the intestinal microbiota has greatly expanded over the last decade, and growing evidence sug- gests that alterations of the intestinal microbiota are critical pathogenic factors that trigger various polyaetiologi- cal diseases associated with increased intestinal permeability and chronic infammation1–3. -

( 12 ) United States Patent
US009956282B2 (12 ) United States Patent ( 10 ) Patent No. : US 9 ,956 , 282 B2 Cook et al. (45 ) Date of Patent: May 1 , 2018 ( 54 ) BACTERIAL COMPOSITIONS AND (58 ) Field of Classification Search METHODS OF USE THEREOF FOR None TREATMENT OF IMMUNE SYSTEM See application file for complete search history . DISORDERS ( 56 ) References Cited (71 ) Applicant : Seres Therapeutics , Inc. , Cambridge , U . S . PATENT DOCUMENTS MA (US ) 3 ,009 , 864 A 11 / 1961 Gordon - Aldterton et al . 3 , 228 , 838 A 1 / 1966 Rinfret (72 ) Inventors : David N . Cook , Brooklyn , NY (US ) ; 3 ,608 ,030 A 11/ 1971 Grant David Arthur Berry , Brookline, MA 4 ,077 , 227 A 3 / 1978 Larson 4 ,205 , 132 A 5 / 1980 Sandine (US ) ; Geoffrey von Maltzahn , Boston , 4 ,655 , 047 A 4 / 1987 Temple MA (US ) ; Matthew R . Henn , 4 ,689 ,226 A 8 / 1987 Nurmi Somerville , MA (US ) ; Han Zhang , 4 ,839 , 281 A 6 / 1989 Gorbach et al. Oakton , VA (US ); Brian Goodman , 5 , 196 , 205 A 3 / 1993 Borody 5 , 425 , 951 A 6 / 1995 Goodrich Boston , MA (US ) 5 ,436 , 002 A 7 / 1995 Payne 5 ,443 , 826 A 8 / 1995 Borody ( 73 ) Assignee : Seres Therapeutics , Inc. , Cambridge , 5 ,599 ,795 A 2 / 1997 McCann 5 . 648 , 206 A 7 / 1997 Goodrich MA (US ) 5 , 951 , 977 A 9 / 1999 Nisbet et al. 5 , 965 , 128 A 10 / 1999 Doyle et al. ( * ) Notice : Subject to any disclaimer , the term of this 6 ,589 , 771 B1 7 /2003 Marshall patent is extended or adjusted under 35 6 , 645 , 530 B1 . 11 /2003 Borody U . -

Outline Release 7 7C
Garrity, et. al., March 6, 2007 Taxonomic Outline of the Bacteria and Archaea, Release 7.7 March 6, 2007. Part 7 – The Bacteria: Phylum “Firmicutes”: Class “Clostridia” George M. Garrity, Timothy G. Lilburn, James R. Cole, Scott H. Harrison, Jean Euzéby, and Brian J. Tindall F Phylum Firmicutes AL N4Lid DOI: 10.1601/nm.3874 Class "Clostridia" N4Lid DOI: 10.1601/nm.3875 71 Order Clostridiales AL Prévot 1953. N4Lid DOI: 10.1601/nm.3876 Family Clostridiaceae AL Pribram 1933. N4Lid DOI: 10.1601/nm.3877 Genus Clostridium AL Prazmowski 1880. GOLD ID: Gi00163. GCAT ID: 000971_GCAT. Entrez genome id: 80. Sequenced strain: BC1 is from a non-type strain. Genome sequencing is incomplete. Number of genomes of this species sequenced 6 (GOLD) 6 (NCBI). N4Lid DOI: 10.1601/nm.3878 Clostridium butyricum AL Prazmowski 1880. Source of type material recommended for DOE sponsored genome sequencing by the JGI: ATCC 19398. High-quality 16S rRNA sequence S000436450 (RDP), M59085 (Genbank). N4Lid DOI: 10.1601/nm.3879 Clostridium aceticum VP (ex Wieringa 1940) Gottschalk and Braun 1981. Source of type material recommended for DOE sponsored genome sequencing by the JGI: ATCC 35044. High-quality 16S rRNA sequence S000016027 (RDP), Y18183 (Genbank). N4Lid DOI: 10.1601/nm.3881 Clostridium acetireducens VP Örlygsson et al. 1996. Source of type material recommended for DOE sponsored genome sequencing by the JGI: DSM 10703. High-quality 16S rRNA sequence S000004716 (RDP), X79862 (Genbank). N4Lid DOI: 10.1601/nm.3882 Clostridium acetobutylicum AL McCoy et al. 1926. Source of type material recommended for DOE sponsored genome sequencing by the JGI: ATCC 824. -

Metatranscriptomic Analysis to Define the Secrebiome, and 16S Rrna
Gallardo‑Becerra et al. Microb Cell Fact (2020) 19:61 https://doi.org/10.1186/s12934‑020‑01319‑y Microbial Cell Factories RESEARCH Open Access Metatranscriptomic analysis to defne the Secrebiome, and 16S rRNA profling of the gut microbiome in obesity and metabolic syndrome of Mexican children Luigui Gallardo‑Becerra1†, Fernanda Cornejo‑Granados1†, Rodrigo García‑López1†, Alejandra Valdez‑Lara1, Shirley Bikel1, Samuel Canizales‑Quinteros2, Blanca E. López‑Contreras2, Alfredo Mendoza‑Vargas3, Henrik Nielsen4 and Adrián Ochoa‑Leyva1* Abstract Background: In the last decade, increasing evidence has shown that changes in human gut microbiota are asso‑ ciated with diseases, such as obesity. The excreted/secreted proteins (secretome) of the gut microbiota afect the microbial composition, altering its colonization and persistence. Furthermore, it infuences microbiota‑host interac‑ tions by triggering infammatory reactions and modulating the host’s immune response. The metatranscriptome is essential to elucidate which genes are expressed under diseases. In this regard, little is known about the expressed secretome in the microbiome. Here, we use a metatranscriptomic approach to delineate the secretome of the gut microbiome of Mexican children with normal weight (NW) obesity (O) and obesity with metabolic syndrome (OMS). Additionally, we performed the 16S rRNA profling of the gut microbiota. Results: Out of the 115,712 metatranscriptome genes that codifed for proteins, 30,024 (26%) were predicted to be secreted, constituting the Secrebiome of the -

Depression and Microbiome—Study on the Relation and Contiguity Between Dogs and Humans
applied sciences Article Depression and Microbiome—Study on the Relation and Contiguity between Dogs and Humans Elisabetta Mondo 1,*, Alessandra De Cesare 1, Gerardo Manfreda 2, Claudia Sala 3 , Giuseppe Cascio 1, Pier Attilio Accorsi 1, Giovanna Marliani 1 and Massimo Cocchi 1 1 Department of Veterinary Medical Science, University of Bologna, Via Tolara di Sopra 50, 40064 Ozzano Emilia, Italy; [email protected] (A.D.C.); [email protected] (G.C.); [email protected] (P.A.A.); [email protected] (G.M.); [email protected] (M.C.) 2 Department of Agricultural and Food Sciences, University of Bologna, Via del Florio 2, 40064 Ozzano Emilia, Italy; [email protected] 3 Department of Physics and Astronomy, Alma Mater Studiorum, University of Bologna, 40126 Bologna, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-051-209-7329 Received: 22 November 2019; Accepted: 7 January 2020; Published: 13 January 2020 Abstract: Behavioral studies demonstrate that not only humans, but all other animals including dogs, can suffer from depression. A quantitative molecular evaluation of fatty acids in human and animal platelets has already evidenced similarities between people suffering from depression and German Shepherds, suggesting that domestication has led dogs to be similar to humans. In order to verify whether humans and dogs suffering from similar pathologies also share similar microorganisms at the intestinal level, in this study the gut-microbiota composition of 12 German Shepherds was compared to that of 15 dogs belonging to mixed breeds which do not suffer from depression. Moreover, the relation between the microbiota of the German Shepherd’s group and that of patients with depression has been investigated. -

Updates on the Sporulation Process in Clostridium Species
Updates on the sporulation process in Clostridium species Talukdar, P. K., Olguín-Araneda, V., Alnoman, M., Paredes-Sabja, D., & Sarker, M. R. (2015). Updates on the sporulation process in Clostridium species. Research in Microbiology, 166(4), 225-235. doi:10.1016/j.resmic.2014.12.001 10.1016/j.resmic.2014.12.001 Elsevier Accepted Manuscript http://cdss.library.oregonstate.edu/sa-termsofuse *Manuscript 1 Review article for publication in special issue: Genetics of toxigenic Clostridia 2 3 Updates on the sporulation process in Clostridium species 4 5 Prabhat K. Talukdar1, 2, Valeria Olguín-Araneda3, Maryam Alnoman1, 2, Daniel Paredes-Sabja1, 3, 6 Mahfuzur R. Sarker1, 2. 7 8 1Department of Biomedical Sciences, College of Veterinary Medicine and 2Department of 9 Microbiology, College of Science, Oregon State University, Corvallis, OR. U.S.A; 3Laboratorio 10 de Mecanismos de Patogénesis Bacteriana, Departamento de Ciencias Biológicas, Facultad de 11 Ciencias Biológicas, Universidad Andrés Bello, Santiago, Chile. 12 13 14 Running Title: Clostridium spore formation. 15 16 17 Key Words: Clostridium, spores, sporulation, Spo0A, sigma factors 18 19 20 Corresponding author: Dr. Mahfuzur Sarker, Department of Biomedical Sciences, College of 21 Veterinary Medicine, Oregon State University, 216 Dryden Hall, Corvallis, OR 97331. Tel: 541- 22 737-6918; Fax: 541-737-2730; e-mail: [email protected] 23 1 24 25 Abstract 26 Sporulation is an important strategy for certain bacterial species within the phylum Firmicutes to 27 survive longer periods of time in adverse conditions. All spore-forming bacteria have two phases 28 in their life; the vegetative form, where they can maintain all metabolic activities and replicate to 29 increase numbers, and the spore form, where no metabolic activities exist. -

Mycobacterium Avium Subspecies Paratuberculosis Infection Modifies Gut Microbiota Under Different Dietary Conditions in a Rabbit
fmicb-07-00446 March 31, 2016 Time: 14:57 # 1 ORIGINAL RESEARCH published: 31 March 2016 doi: 10.3389/fmicb.2016.00446 Mycobacterium avium Subspecies paratuberculosis Infection Modifies Gut Microbiota under Different Dietary Conditions in a Rabbit Model Rakel Arrazuria1, Natalia Elguezabal1*, Ramon A. Juste1, Hooman Derakhshani2 and Ehsan Khafipour2,3* 1 Department of Animal Health, NEIKER-Instituto Vasco de Investigación y Desarrollo Agrario, Derio, Spain, 2 Department of Animal Science, University of Manitoba, Winnipeg, MB, Canada, 3 Department of Medical Microbiology, University of Manitoba, Winnipeg, MB, Canada Mycobacterium avium subspecies paratuberculosis (MAP) the causative agent of paratuberculosis, produces a chronic granulomatous inflammation of the gastrointestinal Edited by: tract of ruminants. It has been recently suggested that MAP infection may be associated Steve Lindemann, with dysbiosis of intestinal microbiota in ruminants. Since diet is one of the key factors Pacific Northwest National affecting the balance of microbial populations in the digestive tract, we intended to Laboratory, USA evaluate the effect of MAP infection in a rabbit model fed a regular or high fiber Reviewed by: Antonis Chatzinotas, diet during challenge. The composition of microbiota of the cecal content and the Helmholtz Centre for Environmental sacculus rotundus was studied in 20 New Zealand white female rabbits. The extracted Research – UFZ, Germany Amélia M. Sarmento, DNA was subjected to paired-end Illumina sequencing of the V3-V4 hypervariable Universidade Fernando Pessoa, region of the 16S rRNA gene for microbiota analysis. Microbial richness (Chao1) Portugal in the cecal content was significantly increased by MAP infection in regular diet *Correspondence: rabbits (p D 0.0043) and marginally increased (p D 0.0503) in the high fiber group.