Chapter 2 Nanoimprint Lithography
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Nanoimprint Lithography: Methods and Material Requirements**
REVIEW DOI: 10.1002/adma.200600882 Nanoimprint Lithography: Methods and Material Requirements** By L. Jay Guo* Nanoimprint lithography (NIL) is a nonconventional lithographic technique for high-throughput patterning of polymer nanostructures at great precision and at low costs. Unlike traditional lithographic ap- proaches, which achieve pattern definition through the use of photons or electrons to modify the chemical and physical properties of the resist, NIL relies on direct mechanical deformation of the resist material and can therefore achieve resolutions beyond the limitations set by light diffraction or beam scattering that are encountered in conventional techniques. This Review covers the basic principles of nanoimprinting, with an emphasis on the requirements on materi- als for the imprinting mold, surface properties, and resist materials for successful and reliable nanostructure replication. 1. Introduction proaches towards nanostructure fabrication have been exploited in the past 15 years, without resorting to expensive The ability to fabricate structures from the micro- to the tools such as those used in deep-UV projection lithography nanoscale with high precision in a wide variety of materials is and electron-beam lithography. These techniques include [1] of crucial importance to the advancement of micro- and nano- microcontact printing (or soft lithography), nanoimprint [2] technology and the nanosciences. The semiconductor industry lithography (NIL), scanning-probe-based techniques (e.g., [3] has been pushing high-precision nanoscale lithography to atomic force microscope lithography), and dip-pen lithogra- [4] manufacture ever-smaller transistors and higher-density inte- phy. A good overview of several of these techniques was [5] grated circuits (ICs). Critical issues, such as resolution, relia- presented in a recent paper. -

Bilayer, Nanoimprint Lithography Brian Faircloth Nuvonyx, Inc., Bridgeton, Missouri 63044 Henry Rohrs Washington University, St
Bilayer, nanoimprint lithography Brian Faircloth Nuvonyx, Inc., Bridgeton, Missouri 63044 Henry Rohrs Washington University, St. Louis, Missouri 63130 Richard Tiberio Cornell University, Ithaca, New York 14853 Rodney Ruoff Washington University, St. Louis, Missouri 63130 Robert R. Krchnaveka) Rowan University, Glassboro, New Jersey 08028 ͑Received 3 May 1999; accepted 21 April 2000͒ Nanoimprint lithography has been shown to be a viable means of patterning polymer films in the sub-100 nm range. In this work, we demonstrate the use of a bilayer resist to facilitate the metal liftoff step in imprinter fabrication. The bilayer resist technology exhibits more uniform patterns and fewer missing features than similar metal nanoparticle arrays fabricated with single layer resist. The bilayer resist relies upon the differential solubility between poly͑methyl methacrylate͒ and poly͑methyl methacrylate methacrylic acid copolymer͒. Evidence is presented that shows the technique has a resolution of better than 10 nm. © 2000 American Vacuum Society. ͓S0734-211X͑00͒03104-8͔ I. INTRODUCTION tion demonstrated in the polymer resist layer. The formation of patterned metal layers is one application. Finely patterned Due to the inevitable transition from the microelectronic metal layers are used as interconnects in integrated circuits. to the nanoelectronic age, the demand for sub-100 nm fea- They can also be used as catalysts for subsequent layer ture sizes in lithographic techniques will increase greatly. As growth. If the subsequent metal layers cannot readily be current devices rapidly approach the 100 nm barrier, the mi- etched, e.g., due to crystalline dependent etching rates, an croelectronics industry is considering several technologies to additive approach such as liftoff is desirable. -

Large-Area Nanoimprint Lithography and Applications
Chapter 3 Large-Area Nanoimprint Lithography and Applications Hongbo Lan Additional information is available at the end of the chapter http://dx.doi.org/10.5772/intechopen.72860 Abstract Large-area nanoimprint lithography (NIL) has been regarded as one of the most prom- ising micro/nano-manufacturing technologies for mass production of large-area micro/ nanoscale patterns and complex 3D structures and high aspect ratio features with low cost, high throughput, and high resolution. That opens the door and paves the way for many commercial applications not previously conceptualized or economically feasible. Great progresses in large-area nanoimprint lithography have been achieved in recent years. This chapter mainly presents a comprehensive review of recent advances in large- area NIL processes. Some promising solutions of large-area NIL and emerging methods, which can implement mass production of micro-and nanostructures over large areas on various substrates or surfaces, are described in detail. Moreover, numerous industrial- level applications and innovative products based on large-area NIL are also demon- strated. Finally, prospects, challenges, and future directions for industrial scale large- area NIL are addressed. An infrastructure of large-area nanoimprint lithography is proposed. In addition, some recent progresses and research activities in large-area NIL suitable for high volume manufacturing environments from our Labs are also intro- duced. This chapter may provide a reference and direction for the further explorations and studies of large-area micro/nanopatterning technologies. Keywords: large-area nanoimprint lithography, large-area micro/nanopatterning, full wafer NIL, roller-type NIL, roll-to-plate NIL, roll-to-roll NIL 1. -

And Nanolithography Techniques and Their Applications
Review on Micro- and Nanolithography Techniques and their Applications Alongkorn Pimpin* and Werayut Srituravanich** Department of Mechanical Engineering, Faculty of Engineering, Chulalongkorn University, Pathumwan, Bangkok 10330, Thailand E-mail: [email protected]*, [email protected]** Abstract. This article reviews major micro- and nanolithography techniques and their applications from commercial micro devices to emerging applications in nanoscale science and engineering. Micro- and nanolithography has been the key technology in manufacturing of integrated circuits and microchips in the semiconductor industry. Such a technology is also sparking revolutionizing advancements in nanotechnology. The lithography techniques including photolithography, electron beam lithography, focused ion beam lithography, soft lithography, nanoimprint lithography and scanning probe lithography are discussed. Furthermore, their applications are summarized into four major areas: electronics and microsystems, medical and biotech, optics and photonics, and environment and energy harvesting. Keywords: Nanolithography, photolithography, electron beam lithography, focused ion beam lithography, soft lithography, nanoimprint lithography, scanning probe lithography, dip-pen lithography, microsystems, MEMS, nanoscience, nanotechnology, nano-engineering. ENGINEERING JOURNAL Volume 16 Issue 1 Received 18 August 2011 Accepted 8 November Published 1 January 2012 Online at http://www.engj.org DOI:10.4186/ej.2012.16.1.37 DOI:10.4186/ej.2012.16.1.37 1. Introduction For decades, micro- and nanolithography technology has been contributed to the manufacturing of integrated circuits (ICs) and microchips. This advance in the semiconductor and IC industry has led to a new paradigm of the information revolution via computers and the internet. Micro- and nanolithography is the technology that is used to create patterns with a feature size ranging from a few nanometers up to tens of millimeters. -
Nano-Lithography
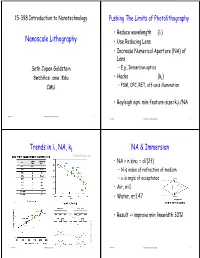
15-398 Introduction to Nanotechnology Pushing The Limits of Photolithography • Reduce wavelength (λ) Nanoscale Lithography • Use Reducing Lens • Increase Numerical Aperture (NA) of Lens Seth Copen Goldstein – E.g., Immersion optics [email protected] •Hacks (k1) CMU – PSM, OPC, RET, off-axis illumination • Rayleigh eqn: min-feature-size=k1λ/NA lecture 4 © 2004-5 Seth Copen Goldstein 1 lecture 4 © 2004-5 Seth Copen Goldstein 2 Trends in λ, NA, k1 NA & Immersion ICKnowledge.com • NA = n sinα = d/(2f) – N is index of refraction of medium – α is angle of acceptance •Air, n=1 • Water, n≅1.47 • Result -> improve min linewidth 30%! CUHG, Chap 5. lecture 4 © 2004-5 Seth Copen Goldstein 3 lecture 4 © 2004-5 Seth Copen Goldstein 4 E-Beam? FIB? Example of E-beam patterning • Use high-energy electrons to alter/ablate a resist P = 40 nm P = 45 nm • Issues: – Secondary electrons – Scattering in resist (or off substrate) – Serial process P = 50 nm P = 60 nm – alignment Handbook of Microlithography, Micromachining and lecture 4 © 2004-5 Seth Copen Goldstein Microfabrication, SPIE (1997)5 lectureUofA, 4 Nanolithography © 2004-5 Seth Copen Goldstein 6 Microelectronics isn’t everything Today • Reactive organics • Soft Lithography/Nanoimprint • 3-D structures • Scanning Probe Lithography • Edge Lithography • Top-down fabrication to create nanoscale features using a physical mold or tip for patterning. lecture 4 © 2004-5 Seth Copen Goldstein 7 lecture 4 © 2004-5 Seth Copen Goldstein 8 Soft Litho Basic Example • Replica Molding transfer features from master to -

Nanolithography
Chapter 8 Nanolithography Gunasekaran Venugopal and Sang-Jae Kim Additional information is available at the end of the chapter http://dx.doi.org/10.5772/55527 1. Introduction Nanolithography is the branch of nanotechnology concerned with the study and application of the nanofabrication of nanometer-scale structures, meaning nanopatterning with at least one lateral dimension between the size of an individual atom and approximately 100 nm. The term nanolithography is derived from the Greek words “nanos”, meaning dwarf; “lithos”, meaning rock or stone; and “graphein” meaning to write. Therefore the literal translation is "tiny writing on stone", however nowadays one understands something different whenever this term is associated with nanotechnology. Nanolithography is used e.g. during the nano‐ fabrication of leading-edge semiconductor integrated circuits (nanocircuitry), for nanoelec‐ tromechanical systems (NEMS) or for almost any other fundamental application across various scientific disciplines in nanoresearch. This technology can be suitable to use in nanofabrication of various semiconducting Integrated Circuits (ICs), NEMS and for various applications in research. The modification in semicon‐ ductor chips at the nano-scale (in the range of 10-9 meter) is also possible. This method is contrasting to various existing nanolithographic techniques like Photolithography (Venugo‐ pal, 2011), Nanoimprint lithography (NIL), Scanning Probe Lithography (SPL), Atomic Force Microscope (AFM) nanolithography, Extreme Ultraviolet Lithography (EUVL) and X-ray Lithography. In this chapter, the various nanolithographic fabrication techniques will be discussed in detail in which we will focus the various nano-patterning techniques/procedures suitable for device fabrication and their engineering applications. This technique is mainly used for nanofabrica‐ tion. -

UV Nanoimprint Lithography: Geometrical Impact on Filling Properties of Nanoscale Patterns
nanomaterials Article UV Nanoimprint Lithography: Geometrical Impact on Filling Properties of Nanoscale Patterns Christine Thanner and Martin Eibelhuber * EV Group, DI Erich Thallner Str. 1, 4782 St. Florian am Inn, Austria; [email protected] * Correspondence: [email protected] Abstract: Ultraviolet (UV) Nanoimprint Lithography (NIL) is a replication method that is well known for its capability to address a wide range of pattern sizes and shapes. It has proven to be an efficient production method for patterning resist layers with features ranging from a few hundred micrometers and down to the nanometer range. Best results can be achieved if the fundamental behavior of the imprint resist and the pattern filling are considered by the equipment and process parameters. In particular, the material properties and pattern size and shape play a crucial role. For capillary force-driven filling behavior it is important to understand the influencing parameters and respective failure modes in order to optimize the processes for reliable full wafer manufacturing. In this work, the nanoimprint results obtained for different pattern geometries are compared with respect to pattern quality and residual layer thickness: The comprehensive overview of the relevant process parameters is helpful for setting up NIL processes for different nanostructures with minimum layer thickness. Keywords: nanoimprint lithography; UV-NIL; SmartNIL Citation: Thanner, C.; Eibelhuber, M. UV Nanoimprint Lithography: Geometrical Impact on Filling Properties of Nanoscale Patterns. 1. Introduction Nanomaterials 2021, 11, 822. Since it was first mentioned in literature [1], nanoimprint lithography (NIL) emerged https://doi.org/10.3390/ into an attractive patterning technique and developed considerably in terms of materials, nano11030822 process technology, and equipment. -

Fabrication of 60-Nm Transistors on 4-In. Wafer Using Nanoimprint at All Lithography Levels Wei Zhanga) and Stephen Y
APPLIED PHYSICS LETTERS VOLUME 83, NUMBER 8 25 AUGUST 2003 Fabrication of 60-nm transistors on 4-in. wafer using nanoimprint at all lithography levels Wei Zhanga) and Stephen Y. Chou NanoStructure Laboratory, Department of Electrical Engineering, Princeton University, Princeton, New Jersey 08544 ͑Received 28 March 2003; accepted 10 June 2003͒ Nanoimprint lithography ͑NIL͒ is a paradigm-shift method that has shown sub-10-nm resolution, high throughput, and low cost. To make NIL a next-generation lithography tool to replace conventional lithography, one must demonstrate the needed overlay accuracy in multilayer NIL, large-area uniformity, and low defect density. Here, we present the fabrication of 60-nm channel metal–oxide–semiconductor field-effect transistors on whole 4-in. wafers using NIL at all lithography levels. The nanotransistors exhibit excellent operational characteristics across the wafer. The statistics from consecutive multiwafer processing show an average overlay accuracy of 500 nm over the entire 4-in. wafer. The accuracy is much better when the field size is reduced. The overlay accuracies are limited by the current alignment method and can be improved substantially. The work presents a significant advance in nanoimprint development and its applications in manufacturing of integrated electrical, optical, chemical, and biological nanocircuits. © 2003 American Institute of Physics. ͓DOI: 10.1063/1.1600505͔ Lithography, a key step in defining the size of micro/ can be a next-generation lithography to replace conventional nanodevices, is a critical tool for microchip manufacturing. lithography in IC manufacturing, NIL must demonstrate the Conventional lithography creates features based on the prin- needed overlay accuracy, large-area uniformity, and low de- ciple of using a radiation ͑e.g., light͒ to locally modify the fect density. -

Soft Thermal Nanoimprint Lithography Using a Nanocomposite Mold
Nano Research https://doi.org/10.1007/s12274-017-1900-0 Soft thermal nanoimprint lithography using a nanocomposite mold Viraj Bhingardive, Liran Menahem, and Mark Schvartzman () Department of Materials Engineering, Isle Katz Institute of Nanoscale Science and Technology, Ben-Gurion University of the Negev, P.O. Box 653, Beer-Sheva 8410501, Israel Received: 6 September 2017 ABSTRACT Revised: 15 October 2017 Soft nanoimprint lithography has been limited to ultraviolet (UV) curable resists. Accepted: 25 October 2017 Here, we introduce a novel approach for soft thermal nanoimprinting. This unprecedented combination of the terms “soft” and “thermal” for nanoimprinting © Tsinghua University Press became possible thanks to an innovative nanocomposite mold consisting of a and Springer-Verlag GmbH flexible polydimethylsiloxane (PDMS) substrate with chemically attached rigid relief features. We used soft thermal nanoimprinting to produce high-resolution Germany 2017 nanopatterns with a sub-100 nm feature size. Furthermore, we demonstrate the applicability of our nanoimprint approach for the nanofabrication of thermally KEYWORDS imprinted nanopatterns on non-planar surfaces such as lenses. Our new soft lithography, nanofabrication strategy paves the way to numerous applications that require nanoimprint lithography, the direct fabrication of functional nanostructures on unconventional substrates. PDMS, non-planar substrates 1 Introduction nanopatterns produced by PDMS molds are practically free of defects. In addition, nanoimprint with soft Soft nanoimprinting is a versatile, high-throughput, molds does not require high pressure; it can be and cost-effective nanolithography method in which performed by gentle pressing with the thumb [6]. a nanoscale pattern is mechanically transferred onto Finally, flexible PDMS molds can be applied to a resist by an elastomeric mold. -

Nanofabrication Techniques for Controlled Drug Release Devices
Nanofabrication Techniques for Controlled Drug Release Devices Lei Chen*, Gerard Henein, and Vincent Luciani Center for Nanoscale Science and Technology National Institute of Standards and Technology 100 Bureau Drive, Stop 620-1, Gaithersburg, MD 20899-6201, USA *Corresponding Author Lei Chen Center for Nanoscale Science and Technology Nantional Institute of Standards and Technology 100 Bureau Drive, Stop 620-1, Gaithersburg, MD 20899-6201, USA Phone: 301-975-2908 Fax: 301-975-8761 Email: [email protected] Gerard Henein Center for Nanoscale Science and Technology Nantional Institute of Standards and Technology 100 Bureau Drive, Stop 620-1, Gaithersburg, MD 20899-6201, USA Vincent Luciani Center for Nanoscale Science and Technology Nantional Institute of Standards and Technology 100 Bureau Drive, Stop 620-1, Gaithersburg, MD 20899-6201, USA New drugs and delivery systems have undergone a rapid development in recent years for treating cancer, HIV aids, diseased organs, damaged nerves to relieve pain, preventing disease and restoring health to human beings (1). The application of nanotechnology in nanomedicne for drug delivery is a new field and has raised great attention. Compared to conventional drug delivery mechanisms, nano-scale systems can deliver the drugs to diseased cells directly and minimize damage to healthy cells, and they can also be made small enough to be implantable in the body. Alternatively, by modifying the shells of the drug, shells can release the drug automatically under certain stimuli. Nanoscale controlled-release drug systems provide highly selective and effective therapeutic modalities (2). Controlled-Release Drug Delivery Vehicles The first generation nanotechnology-based controlled-released drug products appeared around 2005. -

Introduction to Nanolithography Introduction to Nanolithography
Introduction to Nanolithography Junwei Su Mechanical Engineering University of Massachusetts Lowell [email protected] Outline • Introduction • Photolithography • SfSoft Lihithograp hy • Nanoimprint Lithography (NIL) • NIL System Overview • Conclusion Nanolithography Nanolithography is the branch of nanotechnology concerned with the stdtudy and applica tion of fbifabrica ting nanometer‐scale structures, meaning patterns with at least one lateral dimension between the size of an individual atom and approximately 100 nm. Introduction • Why we need micro/nano pattern? MEMS (Micro electronic mechanical system) A MEMS device optical switch ratcheted microgear [Sandia National Laboratories] (Sandia) (Sandia) Top‐down Versus Bottom‐up Top Down Process Bottom Up Process Start with bulk Start with bulk wafer wafer Apply layer of Alteeaeaor area of w aeafer photoresist where structure is to be created by adding polymer or seed Expose wafer with crystals or other UV light through techniques. mask and etch Grow or assemble wafer the structure on the area determined by Etched wafer with the seed crystals or desired pattern polymer. (self assembly) Similar results can be obtained through bottom-up and top-down processes Photolithography https://www.youtube.com/watch?v=1bxf9QRVesQ EUV lithography • Extreme ultraviolet lithography (also known as EUV or EUVL) is a next‐ generation lithography technology using an EUV wavelength, currently expected to be 13.5 nm. Photolithography Limits • It requir es epesexpensive instr um en ts adand facili ti es with high capital investment • Pattern resolution is limited by optical diffraction • The methods is not suitable for patterning all types of polymers, and only photosensitive resist materials can be directly patterned. • The technique requires harsh processing conditions. -

Mastering of NIL Stamps with Undercut T-Shaped Features from Single Layer to Multilayer Stamps
nanomaterials Article Mastering of NIL Stamps with Undercut T-Shaped Features from Single Layer to Multilayer Stamps Philipp Taus 1, Adrian Prinz 2, Heinz D. Wanzenboeck 1,*, Patrick Schuller 1, Anton Tsenov 1, Markus Schinnerl 1, Mostafa M. Shawrav 3, Michael Haslinger 4 and Michael Muehlberger 4 1 TU Wien, Institute for Solid State Electronics, 1040 Vienna, Austria; [email protected] (P.T.); [email protected] (P.S.); [email protected] (A.T.); [email protected] (M.S.) 2 Stratec Consumables GmbH, 5081 Anif, Austria; [email protected] 3 ams AG, Tobelbader Strasse 30, 8141 Premstaetten, Austria; [email protected] 4 PROFACTOR GmbH, 4407 Steyr, Austria; [email protected] (M.H.); [email protected] (M.M.) * Correspondence: [email protected]; Tel.: +43-1-588-36243 Abstract: Biomimetic structures such as structural colors demand a fabrication technology of complex three-dimensional nanostructures on large areas. Nanoimprint lithography (NIL) is capable of large area replication of three-dimensional structures, but the master stamp fabrication is often a bottleneck. We have demonstrated different approaches allowing for the generation of sophisticated undercut T-shaped masters for NIL replication. With a layer-stack of phase transition material (PTM) on poly-Si, we have demonstrated the successful fabrication of a single layer undercut T-shaped structure. With a multilayer-stack of silicon oxide on silicon, we have shown the successful fabrication of a multilayer Citation: Taus, P.; Prinz, A.; undercut T-shaped structures. For patterning optical lithography, electron beam lithography and Wanzenboeck, H.D.; Schuller, P.; nanoimprint lithography have been compared and have yielded structures from 10 µm down to Tsenov, A.; Schinnerl, M.; Shawrav, 300 nm.