Systematic Discovery and Characterization of Regulatory Motifs in ENCODE TF Binding Experiments
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Compensation Among HMGN Variants Modulates the Dnase I Hypersensitive Sites at Enhancers
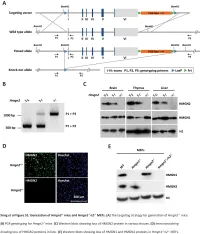
Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press Research Functional compensation among HMGN variants modulates the DNase I hypersensitive sites at enhancers Tao Deng,1,12 Z. Iris Zhu,2,12 Shaofei Zhang,1 Yuri Postnikov,1 Di Huang,2 Marion Horsch,3 Takashi Furusawa,1 Johannes Beckers,3,4,5 Jan Rozman,3,5 Martin Klingenspor,6,7 Oana Amarie,3,8 Jochen Graw,3,8 Birgit Rathkolb,3,5,9 Eckhard Wolf,9 Thure Adler,3 Dirk H. Busch,10 Valérie Gailus-Durner,3 Helmut Fuchs,3 Martin Hrabeˇ de Angelis,3,4,5 Arjan van der Velde,2,13 Lino Tessarollo,11 Ivan Ovcherenko,2 David Landsman,2 and Michael Bustin1 1Protein Section, Laboratory of Metabolism, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA; 2Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, Bethesda, Maryland 20892, USA; 3German Mouse Clinic, Institute of Experimental Genetics, Helmholtz Zentrum München, German Research Center for Environmental Health, 85764 Neuherberg, Germany; 4Experimental Genetics, Center of Life and Food Sciences Weihenstephan, Technische Universität München, 85354 Freising-Weihenstephan, Germany; 5German Center for Diabetes Research (DZD), 85764 Neuherberg, Germany; 6Molecular Nutritional Medicine, Technische Universität München, 85350 Freising, Germany; 7Center for Nutrition and Food Sciences, Technische Universität München, 85350 Freising, Germany; 8Institute of Developmental Genetics (IDG), 85764 -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

HMGB1 in Health and Disease R
Donald and Barbara Zucker School of Medicine Journal Articles Academic Works 2014 HMGB1 in health and disease R. Kang R. C. Chen Q. H. Zhang W. Hou S. Wu See next page for additional authors Follow this and additional works at: https://academicworks.medicine.hofstra.edu/articles Part of the Emergency Medicine Commons Recommended Citation Kang R, Chen R, Zhang Q, Hou W, Wu S, Fan X, Yan Z, Sun X, Wang H, Tang D, . HMGB1 in health and disease. 2014 Jan 01; 40():Article 533 [ p.]. Available from: https://academicworks.medicine.hofstra.edu/articles/533. Free full text article. This Article is brought to you for free and open access by Donald and Barbara Zucker School of Medicine Academic Works. It has been accepted for inclusion in Journal Articles by an authorized administrator of Donald and Barbara Zucker School of Medicine Academic Works. Authors R. Kang, R. C. Chen, Q. H. Zhang, W. Hou, S. Wu, X. G. Fan, Z. W. Yan, X. F. Sun, H. C. Wang, D. L. Tang, and +8 additional authors This article is available at Donald and Barbara Zucker School of Medicine Academic Works: https://academicworks.medicine.hofstra.edu/articles/533 NIH Public Access Author Manuscript Mol Aspects Med. Author manuscript; available in PMC 2015 December 01. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Mol Aspects Med. 2014 December ; 0: 1–116. doi:10.1016/j.mam.2014.05.001. HMGB1 in Health and Disease Rui Kang1,*, Ruochan Chen1, Qiuhong Zhang1, Wen Hou1, Sha Wu1, Lizhi Cao2, Jin Huang3, Yan Yu2, Xue-gong Fan4, Zhengwen Yan1,5, Xiaofang Sun6, Haichao Wang7, Qingde Wang1, Allan Tsung1, Timothy R. -

Hepatitis C Virus As a Unique Human Model Disease to Define
viruses Review Hepatitis C Virus as a Unique Human Model Disease to Define Differences in the Transcriptional Landscape of T Cells in Acute versus Chronic Infection David Wolski and Georg M. Lauer * Liver Center at the Gastrointestinal Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA * Correspondence: [email protected]; Tel.: +1-617-724-7515 Received: 27 June 2019; Accepted: 23 July 2019; Published: 26 July 2019 Abstract: The hepatitis C virus is unique among chronic viral infections in that an acute outcome with complete viral elimination is observed in a minority of infected patients. This unique feature allows direct comparison of successful immune responses with those that fail in the setting of the same human infection. Here we review how this scenario can be used to achieve better understanding of transcriptional regulation of T-cell differentiation. Specifically, we discuss results from a study comparing transcriptional profiles of hepatitis C virus (HCV)-specific CD8 T-cells during early HCV infection between patients that do and do not control and eliminate HCV. Identification of early gene expression differences in key T-cell differentiation molecules as well as clearly distinct transcriptional networks related to cell metabolism and nucleosomal regulation reveal novel insights into the development of exhausted and memory T-cells. With additional transcriptional studies of HCV-specific CD4 and CD8 T-cells in different stages of infection currently underway, we expect HCV infection to become a valuable model disease to study human immunity to viruses. Keywords: viral hepatitis; hepatitis C virus; T cells; transcriptional regulation; transcription factors; metabolism; nucleosome 1. -
Supplemental Data.Pdf
Table S1. Summary of sequencing results A. DNase‐seq data % Align % Mismatch % >=Q30 bases Sample ID # Reads % unique reads (PF) Rate (PF) (PF) WT_1 84,301,522 86.76 0.52 93.34 96.30 WT_2 98,744,222 84.97 0.51 93.75 89.94 Hmgn1‐/‐_1 79,620,656 83.94 0.86 90.58 88.65 Hmgn1‐/‐_2 62,673,782 84.13 0.87 91.30 89.18 Hmgn2‐/‐_1 87,734,440 83.49 0.71 91.81 90.00 Hmgn2‐/‐_2 82,498,808 83.25 0.69 92.73 90.66 Hmgn1‐/‐n2‐/‐_1 71,739,638 68.51 2.31 81.11 89.22 Hmgn1‐/‐n2‐/‐_2 74,113,682 68.19 2.37 81.16 86.57 B. ChIP‐seq data Histone % Align % Mismatch % >=Q30 % unique Genotypes # Reads marks (PF) Rate (PF) bases (PF) reads H3K4me1 100670054 92.99 0.28 91.21 87.29 H3K4me3 67064272 91.97 0.35 89.11 27.15 WT H3K27ac 90,340,242 93.57 0.28 95.02 89.80 input 111,292,572 78.24 0.55 96.07 86.99 H3K4me1 84598176 92.34 0.33 91.2 81.69 H3K4me3 90032064 92.19 0.44 88.76 15.81 Hmgn1‐/‐n2‐/‐ H3K27ac 86,260,526 93.40 0.29 94.94 87.49 input 78,142,334 78.47 0.56 95.82 81.11 C. MNase‐seq data % Mismatch % >=Q30 bases % unique Sample ID # Reads % Align (PF) Rate (PF) (PF) reads WT_1_Extensive 45,232,694 55.23 1.49 90.22 81.73 WT_1_Limited 105,460,950 58.03 1.39 90.81 79.62 WT_2_Extensive 40,785,338 67.34 1.06 89.76 89.60 WT_2_Limited 105,738,078 68.34 1.05 90.29 85.96 Hmgn1‐/‐n2‐/‐_1_Extensive 117,927,050 55.74 1.49 89.50 78.01 Hmgn1‐/‐n2‐/‐_1_Limited 61,846,742 63.76 1.22 90.57 84.55 Hmgn1‐/‐n2‐/‐_2_Extensive 137,673,830 60.04 1.30 89.28 78.99 Hmgn1‐/‐n2‐/‐_2_Limited 45,696,614 62.70 1.21 90.71 85.52 D. -

Identification of a Proteomic Signature of Senescence in Primary Human
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.22.309351; this version posted January 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Identification of a Proteomic Signature of Senescence in Primary Human Mammary Epithelial Cells Alireza Delfarah1†, DongQing Zheng1, Jesse Yang1 and Nicholas A. Graham1,2,3 1 Mork Family Department of Chemical Engineering and Materials Science, 2 Norris Comprehensive Cancer Center, 3 Leonard Davis School of Gerontology, University of Southern California, Los Angeles, CA 90089 †Current address, Department of Genetics, Stanford University, Palo Alto, CA 94305 Running title: Proteomic profiling of primary HMEC senescence Keywords: replicative senescence, aging, mammary epithelial cell, proteomics, data integration, transcriptomics Corresponding author: Nicholas A. Graham, University of Southern California, 3710 McClintock Ave., RTH 509, Los Angeles, CA 90089. Phone: 213-240-0449; E-mail: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.09.22.309351; this version posted January 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Proteomic profiling of primary HMEC senescence Abstract Senescence is a permanent cell cycle arrest that occurs in response to cellular stress. Because senescent cells promote age-related disease, there has been considerable interest in defining the proteomic alterations in senescent cells. Because senescence differs greatly depending on cell type and senescence inducer, continued progress in the characterization of senescent cells is needed. Here, we analyzed primary human mammary epithelial cells (HMECs), a model system for aging, using mass spectrometry-based proteomics. -

Virtual Chip-Seq: Predicting Transcription Factor Binding
bioRxiv preprint doi: https://doi.org/10.1101/168419; this version posted March 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Virtual ChIP-seq: predicting transcription factor binding 2 by learning from the transcriptome 1,2,3 1,2,3,4,5 3 Mehran Karimzadeh and Michael M. Hoffman 1 4 Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada 2 5 Princess Margaret Cancer Centre, Toronto, ON, Canada 3 6 Vector Institute, Toronto, ON, Canada 4 7 Department of Computer Science, University of Toronto, Toronto, ON, Canada 5 8 Lead contact: michael.hoff[email protected] 9 March 8, 2019 10 Abstract 11 Motivation: 12 Identifying transcription factor binding sites is the first step in pinpointing non-coding mutations 13 that disrupt the regulatory function of transcription factors and promote disease. ChIP-seq is 14 the most common method for identifying binding sites, but performing it on patient samples is 15 hampered by the amount of available biological material and the cost of the experiment. Existing 16 methods for computational prediction of regulatory elements primarily predict binding in genomic 17 regions with sequence similarity to known transcription factor sequence preferences. This has limited 18 efficacy since most binding sites do not resemble known transcription factor sequence motifs, and 19 many transcription factors are not even sequence-specific. 20 Results: 21 We developed Virtual ChIP-seq, which predicts binding of individual transcription factors in new 22 cell types using an artificial neural network that integrates ChIP-seq results from other cell types 23 and chromatin accessibility data in the new cell type. -

NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 11-7-2018 Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer Jairo Ramos [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Clinical Epidemiology Commons Recommended Citation Ramos, Jairo, "Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer" (2018). FIU Electronic Theses and Dissertations. 3872. https://digitalcommons.fiu.edu/etd/3872 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida DECIPHER MECHANISMS BY WHICH NUCLEAR RESPIRATORY FACTOR ONE (NRF1) COORDINATES CHANGES IN THE TRANSCRIPTIONAL AND CHROMATIN LANDSCAPE AFFECTING DEVELOPMENT AND PROGRESSION OF INVASIVE BREAST CANCER A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in PUBLIC HEALTH by Jairo Ramos 2018 To: Dean Tomás R. Guilarte Robert Stempel College of Public Health and Social Work This dissertation, Written by Jairo Ramos, and entitled Decipher Mechanisms by Which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer, having been approved in respect to style and intellectual content, is referred to you for judgment. -

Binding RXFP1 in Brain Tumors
Functional role of C1Q-TNF related peptide 8 (CTRP8)- binding RXFP1 in brain tumors By THATCHAWAN THANASUPAWAT A Thesis submitted to the Faculty of Graduate Studies of The University of Manitoba in partial fulfillment of the requirements of the degree of DOCTOR OF PHILOSOPHY Department of Human Anatomy and Cell Science University of Manitoba Winnipeg, Manitoba Canada Copyright © 2017 by Thatchawan Thanasupawat TABLE OF CONTENTS ABSTRACT ....................................................................................................................... v ACKNOWLEDGEMENTS ........................................................................................... vii LIST OF TABLES ........................................................................................................... ix LIST OF FIGURES .......................................................................................................... x LIST OF ABBREVIATIONS ........................................................................................ xii LIST OF COPYRIGHTED MATERIALS ................................................................. xix CHAPTER 1: INTRODUCTION .................................................................................... 1 1.1 Relaxin family peptide receptors (RXFPs) ............................................................... 1 1.2 Relaxin family peptide receptor 1 (RXFP1) ............................................................. 1 1.2.1 Structural features of RXFP1 ............................................................................ -

Identification of Proteins Involved in the Maintenance of Genome Stability
Identification of Proteins Involved in the Maintenance of Genome Stability by Edith Hang Yu Cheng A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Biochemistry University of Toronto ©Copyright by Edith Cheng2015 Identification of Proteins Involved in the Maintenance of Genome Stability Edith Cheng Doctor of Philosophy Department of Biochemistry University of Toronto 2015 Abstract Aberrant changes to the genome structure underlie numerous human diseases such as cancers. The functional characterization ofgenesand proteins that maintain chromosome stability will be important in understanding disease etiology and developing therapeutics. I took a multi-faceted approach to identify and characterize genes involved in the maintenance of genome stability. As biological pathways involved in genome maintenance are highly conserved in evolution, results from model organisms can greatly facilitate functional discovery in humans. In S. cerevisiae, I identified 47 essential gene depletions with elevated levels of spontaneous DNA damage foci and 92 depletions that caused elevated levels of chromosome rearrangements. Of these, a core subset of 15 DNA replication genes demonstrated both phenotypes when depleted. Analysis of rearrangement breakpoints revealed enrichment at yeast fragile sites, Ty retrotransposons, early origins of replication and replication termination sites. Together, thishighlighted the integral role of DNA replicationin genome maintenance. In light of my findings in S. cerevisiae, I identified a list of 153 human proteins that interact with the nascentDNA at replication forks, using a DNA pull down strategy (iPOND) in human cell lines. As a complementary approach for identifying human proteins involved in genome ii maintenance, I usedthe BioID techniqueto discernin vivo proteins proximal to the human BLM- TOP3A-RMI1-RMI2 genome stability complex, which has an emerging role in DNA replication progression. -

The Hmgn Family of Chromatin Binding Proteins Regulate Stem Cell Pluripotency and Neuronal Differentiation
The Hmgn family of chromatin binding proteins regulate stem cell pluripotency and neuronal differentiation Gokula Mohan, Ohoud Rehbini, Tomoko Iwata, Katherine West Institute of Cancer Sciences, University of Glasgow Hmgn proteins Hmgn proteins are enriched across Hmgn2 is expressed in neural progenitor cells the Oct4 and Nanog gene loci during embryogenesis E11.5 E13.5 E15.5 Bind as dimers to nucleosomes svz Hmgn1 Hmgn2 svz 3 Regulate the deposition of histone 3 3 2.5 modifications 2.5 2.5 2 2 2 1.5 Compete with linker histones 1.5 1.5 1 enrichment 1 1 Interact with several transcription 0.5 0.5 0.5 factors. 0 0 0 svz: ventricular/ -1781 -399 410 2070 3342 -1081 -219 929 1740 2636 Actin Msat c subventricular zone Kato H et al. PNAS 2011;108:12283-12288 Oct4 Nanog H3 dg dg: dentate gyrus (in hippocampus) control cells NLS Nucleosome-binding domain NLS Regulatory domain 2 2 2 very highly conserved Competes with H1 c: cerebellum 1.5 1.5 1.5 100 aa 1 1 1 P14 Inhibits phosphorylation Increases acetylation enrichment 0.5 0.5 0.5 of H3S10 of H3K14 0 0 0 -1781 -399 410 2070 3342 -1081 -219 929 1740 2636 Actin Msat Retinoic acid-induced differentiation of EC P Oct4 Nanog cells down the neuronal lineage. M A A P Reprogramming Differentiation Hmgn1 and Hmgn2 binding profiles in control cells. Enrichment at each PCR set was normalised to Histone H3 N-terminal tail ARTKQTARKSTGGKAPRKQLATKAARKSA... stage stage input and then normalised to average H3 enrichment across all primer sets to control for differences M between chromatin preps.