Models of Gene Gain and Gene Loss for Probabilistic Reconstruction of Gene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Arts & Letters of Rocky Neck in the 1950S
GLOUCESTER, MASSACHUSETTS TheArts & Letters of Rocky Neck in the 1950s by Martha Oaks have received the attention they deserve. Four Winds: The Arts & Letters of Rocky With this exhibition, Four Winds, the Cape Neck in the 1950s is on view through Sep- ocky Neck holds the distinction as Ann Museum casts the spotlight on one of tember 29, 2013, at the Cape Ann Mu- R one of the most important places in those interludes: the decade and a half fol- seum, 27 Pleasant Street, Gloucester, American art history. Since the mid-nine- lowing the Second World War. Not so far Massachusetts, 01930, 978-283-0455, teeth century, its name has been associated in the past that it cannot be recollected by www.capeannmuseum.org. A 52-page soft with many of this country’s best known many, yet just far enough that it is apt to cover catalogue accompanies the exhibition. artists: Winslow Homer, Frank Duveneck, be lost, the late 1940s and 1950s found a All illustrated works are from the Cape Theresa Bernstein, Jane Peterson and Ed- young and vibrant group of artists working Ann Museum unless otherwise noted. ward Hopper. From the mid-1800s on the Neck. through the first quarter of the twentieth Although it is one of Cape Ann’s painter William Morris Hunt and his pro- century, the heyday of the art colony on longest-lived and best known art colonies, tégé, Helen Mary Knowlton. Hunt was Rocky Neck, the neighborhood was awash Rocky Neck was not the first. One of the one of the first art teachers to welcome with artists. -

Milwaukee County Historical Society
Title: White Family Collection Manuscript Number: Mss-3325 Inclusive Dates: ca. 1925-2009 Quantity: 14.4 cu. ft. Location: WHW, Sh. B004-B006 (14.0 cu. ft.) RC21A, Sh. 005 (0.4 cu. ft.) Abstract: The White Family consisted of husband and wife Joseph Charles White and Nancy Metz White, and their twin daughters Michele and Jacqueline. Nancy was a local artist who designed and created sculptures constructed out of discarded scrap metal, heating and cooling ventilation pipes, and other recycled items. Originally from Madison, she graduated from UW- Madison with a bachelor’s degree in art education and also did graduate study there. She is primarily noted for creating large-scale outdoor public sculptures, which include Tree of Life in Mitchell Boulevard Park in 2002, Magic Grove in Enderis Park in 2006, Helping Hands at Mead Public Library in Sheboygan, and Fantasy Garden at St. John’s On the Lake. In addition to being a sculptor, Nancy also was an art teacher and the Creative Art Coordinator at Urban Day School Elementary from 1970 to 1978. Joseph C. White was born in 1925 in Michigan. He earned bachelor’s and master’s degrees from Northwestern University and also served in the Navy during World War II and the Korean conflict. In the 1960s, as Vice President of Inland Steel Products Company, he led the company’s involvement in the pioneering School Construction Systems Development (SCSD) project for California schools. He left Inland Steel and formed his own company, Syncon, to focus on modular construction projects. He was also an adjunct architecture professor at UW- Milwaukee. -

P S Y C H O S O C I a L W E L L B E I N G S E R I
PSYCHOSOCIAL WELLBEING SERIES Tree of Life A workshop methodology for children, young people and adults Adapted by Catholic Relief Services with permission from REPSSI Third Edition for a Global Audience 1 REPSSI is a non-profit organisation working to lessen the devastating social and Catholic Relief Services (CRS) was founded in 1943 by the Catholic Bishops of emotional (psychosocial) impact of poverty, conflict, HIV and AIDS among children the United States to serve World War II survivors in Europe. Since then, we have and youth. It is led by Noreen Masiiwa Huni, Chief Executive Officer. REPSSI’s aim is expanded in size to reach 100 million people annually in over 100 countries on five to ensure that all children have access to stable care and protection through quality continents. psychosocial support. We work at the international, regional and national level in East and Southern Africa. Our mission is to assist impoverished and disadvantaged people overseas, working in the spirit of Catholic social teaching to promote the sacredness of human life The best way to support vulnerable children and youth is within a healthy family and and the dignity of the human person. Catholic Relief Services works in partnership community environment. We partner with governments, development partners, with local, national and international organizations and structures in emergency international organisations and NGOs to provide programmes that strengthen response, agriculture and health, as well as microfinance, water and sanitation, communities’ and families’ competencies to better promote the psychosocial peace and justice, capacity strengthening, and education. Although our mission wellbeing of their children and youth. -

History That Moves You Forward
HISTORY THAT MOVES YOU FORWARD ANNUAL REPORT 2014 LEADERSHIP The Dallas Holocaust Museum/Center for Education and Tolerance celebrated its 30th CONTENTS anniversary in 2014. We have accomplished so much in the last three decades: moving into our larger space in the West End, preserving the memories of Holocaust survivors and The Dallas Holocaust Museum/Center for Education welcoming more than a million visitors through our doors. and Tolerance is dedicated to preserving the memory Now we enter a new and exciting phase in our history as we prepare to build a permanent facility that will quadruple our space and allow us to accommodate 200,000 visitors annually. of the Holocaust, and to teaching the moral and ethical In 2014, we took a large step forward in reaching this vision through fundraising, expansions in programming and continued attendance growth. More than 65,500 guests visited the response to prejudice, hatred and indifference, for the Museum, representing a 14 percent increase from the previous year. Many of these visitors benefit of all humanity. were drawn by new events such as the Upstander Speaker Series, which brings notable experts from across the world to speak about human rights to our community. We also designed and curated a special exhibit for the first time, ushering in a new era in which our Museum not only teaches the dangers of hate but also contributes to our community's understanding of human rights. Drawn to Action: The Life and Work of Arthur Szyk brought together the works of the great political cartoonist and an analysis of their impact that illustrated how powerful the pen can be in fighting intolerance. -
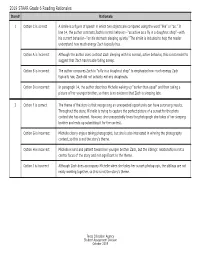
2019 STAAR Grade 6 Reading Rationales Item# Rationale
2019 STAAR Grade 6 Reading Rationales Item# Rationale 1 Option C is correct A simile is a figure of speech in which two objects are compared using the word “like” or “as.” In line 14, the author contrasts Zach’s normal behavior—“as active as a fly in a doughnut shop”—with his current behavior—“on his stomach sleeping quietly.” The simile is included to help the reader understand how much energy Zach typically has. Option A is incorrect Although the author does contrast Zach sleeping with his normal, active behavior, this is not meant to suggest that Zach has trouble falling asleep. Option B is incorrect The author compares Zach to “a fly in a doughnut shop” to emphasize how much energy Zach typically has; Zach did not actually eat any doughnuts. Option D is incorrect In paragraph 14, the author describes Michelle waking up “earlier than usual” and then taking a picture of her younger brother, so there is no evidence that Zach is sleeping late. 2 Option F is correct The theme of the story is that recognizing an unexpected opportunity can have surprising results. Throughout the story, Michelle is trying to capture the perfect picture of a sunset for the photo contest she has entered. However, she unexpectedly loves the photograph she takes of her sleeping brother and ends up submitting it for the contest. Option G is incorrect Michelle clearly enjoys taking photographs, but she is also interested in winning the photography contest, so this is not the story’s theme. Option H is incorrect Michelle is kind and patient toward her younger brother Zach, but the siblings’ relationship is not a central focus of the story and not significant to the theme. -

Days & Hours for Social Distance Walking Visitor Guidelines Lynden
53 22 D 4 21 8 48 9 38 NORTH 41 3 C 33 34 E 32 46 47 24 45 26 28 14 52 37 12 25 11 19 7 36 20 10 35 2 PARKING 40 39 50 6 5 51 15 17 27 1 44 13 30 18 G 29 16 43 23 PARKING F GARDEN 31 EXIT ENTRANCE BROWN DEER ROAD Lynden Sculpture Garden Visitor Guidelines NO CLIMBING ON SCULPTURE 2145 W. Brown Deer Rd. Do not climb on the sculptures. They are works of art, just as you would find in an indoor art Milwaukee, WI 53217 museum, and are subject to the same issues of deterioration – and they endure the vagaries of our harsh climate. Many of the works have already spent nearly half a century outdoors 414-446-8794 and are quite fragile. Please be gentle with our art. LAKES & POND There is no wading, swimming or fishing allowed in the lakes or pond. Please do not throw For virtual tours of the anything into these bodies of water. VEGETATION & WILDLIFE sculpture collection and Please do not pick our flowers, fruits, or grasses, or climb the trees. We want every visitor to be able to enjoy the same views you have experienced. Protect our wildlife: do not feed, temporary installations, chase or touch fish, ducks, geese, frogs, turtles or other wildlife. visit: lynden.tours WEATHER All visitors must come inside immediately if there is any sign of lightning. PETS Pets are not allowed in the Lynden Sculpture Garden except on designated dog days. -

A FAMILY FRIENDLY GUIDE to Services for Milwaukee County Special Needs Children and Youth and Their Families
A FAMILY FRIENDLY GUIDE to Services for Milwaukee County Special Needs Children and Youth and Their Families Project Equal Access for Special Needs Services for Youth INTRODUCTION Milwaukee County offers many programs to support families of children and youth who may have special needs. The child or youth must have a significant disability which is a physical, mental, or emotional limitation which seriously restricts the child’s ability to carry out basic living activities such as self-care, learning, communications, mobility and self-direction. Children and youth from ages birth to 22 can receive support services to help them live at home with their family. Families can access certain services from different agencies throughout the County. We hope this guide will make it easy to locate those services from the different places in Milwaukee County. The guide will provide brief information for each of these programs and how your child can be found eligible for these programs. The agencies that you may connect with include Milwaukee County Disability Services, Milwaukee County BHD Children and Young Adult Community Services and Wraparound Milwaukee, Katie Beckett Program, and the Social Security Administration. When you work with these agencies, you will be asked to provide information about your child such as whether your child is on Medicaid, has a diagnosis from a physician, a psychological evaluation or an individualized education plan (IEP) at school. This Guide is designed as a resource to increase awareness and access to services for Milwaukee African American children with a disability and their families. It is part of DRW’s Project EASSY, Equal Access for Special Needs Services for Youth. -

STAAR® State of Texas Assessments of Academic Readiness
STAAR® State of Texas Assessments of Academic Readiness GRADE 6 Reading May 2019 RELEASED Copyright © 2019, Texas Education Agency. All rights reserved. Reproduction of all or portions of this work is prohibited without express written permission from the Texas Education Agency. STAAR Reading 10/02/2019 G6RSP19R_rev00 STAAR Reading 10/02/2019 G6RSP19R_rev00 READING Reading Page 3 STAAR Reading 10/02/2019 G6RSP19R_rev00 Read the selection and choose the best answer to each question. Then fill in the answer on your answer document. A Picture of Peace 1 When she was just seven years old, Michelle knew with certainty that she wanted to be a photographer when she grew up. That year she received her first camera, a small disposable one to use on the family vacation. At first she randomly clicked the button, not giving much thought to what she was doing. When her father examined her blurred images and aimless shots, he advised Michelle to look through the lens and think about what the resulting picture would look like. The next day Michelle saw a family of ducks, and remembering what her father had said, she lay down on the ground and waited for a duckling to waddle near her. That picture still hangs on her bedroom wall. 2 Now, six years later, Michelle was attempting to capture a sunset for a local photography contest. She groaned as storm clouds rolled in before the sun had a chance to cast its vibrant colors across the sky. 3 “Mom, I don’t think I’m ever going to get this shot!” Michelle complained, putting her camera equipment on the kitchen table and sighing with exasperation. -

Mayor's Office of Arts, Tourism and Special Events Boston Art
Mayor’s Office of Arts, Tourism and Special Events Boston Art Commission 100 Public Artworks: Back Bay, Beacon Hill, the Financial District and the North End 1. Lief Eriksson by Anne Whitney This life-size bronze statue memorializes Lief Eriksson, the Norse explorer believed to be the first European to set foot on North America. Originally sited to overlook the Charles River, Eriksson stands atop a boulder and shields his eyes as if surveying unfamiliar terrain. Two bronze plaques on the sculpture’s base show Eriksson and his crew landing on a rocky shore and, later, sharing the story of their discovery. When Boston philanthropist Eben N. Horsford commissioned the statue, some people believed that Eriksson and his crew landed on the shore of Massachusetts and founded their settlement, called Vinland, here. However, most scholars now consider Vinland to be located on the Canadian coast. This piece was created by a notable Boston sculptor, Anne Whitney. Several of her pieces can be found around the city. Whitney was a fascinating and rebellious figure for her time: not only did she excel in the typically ‘masculine’ medium of large-scale sculpture, she also never married and instead lived with a female partner. 2. Ayer Mansion Mosaics by Louis Comfort Tiffany At first glance, the Ayer Mansion seems to be a typical Back Bay residence. Look more closely, though, and you can see unique elements decorating the mansion’s façade. Both inside and outside, the Ayer Mansion is ornamented with colorful mosaics and windows created by the famed interior designer Louis Comfort Tiffany. -

Origin and History of Seventh-Day Adventists, Vol. 1
Origin and History of Seventh-day Adventists FRONTISPIECE PAINTING BY HARRY ANDERSON © 1949, BY REVIEW AND HERALD As the disciples watched their Master slowly disappear into heaven, they were solemnly reminded of His promise to come again, and of His commission to herald this good news to all the world. Origin and History of Seventh-day Adventists VOLUME ONE by Arthur Whitefield Spalding REVIEW AND HERALD PUBLISHING ASSOCIATION WASHINGTON, D.C. COPYRIGHT © 1961 BY THE REVIEW AND HERALD PUBLISHING ASSOCIATION WASHINGTON, D.C. OFFSET IN THE U.S.A. AUTHOR'S FOREWORD TO FIRST EDITION THIS history, frankly, is written for "believers." The reader is assumed to have not only an interest but a communion. A writer on the history of any cause or group should have suffi- cient objectivity to relate his subject to its environment with- out distortion; but if he is to give life to it, he must be a con- frere. The general public, standing afar off, may desire more detachment in its author; but if it gets this, it gets it at the expense of vision, warmth, and life. There can be, indeed, no absolute objectivity in an expository historian. The painter and interpreter of any great movement must be in sympathy with the spirit and aim of that movement; it must be his cause. What he loses in equipoise he gains in momentum, and bal- ance is more a matter of drive than of teetering. This history of Seventh-day Adventists is written by one who is an Adventist, who believes in the message and mission of Adventists, and who would have everyone to be an Advent- ist. -

Milwaukee Military Memorials: Inventory Sheet General Information Title(S) Subject Matter(S) Current Location Ald. District Stre
Milwaukee Military Memorials: Inventory Sheet Summer 2014 City of Milwaukee Carlen Hatala ([email protected]) City Clerck's Office Paul Jakubovic ([email protected]) Historic Preservation Office Nader Sayadi ([email protected]) 200 E Wells St. Milwaukee, WI (414) 286-5722 General Information Title(s) World War I Memorial Flagpole Property No. Ald.04 - 07 Subject Flagpole Matter(s) Current Ald. District 4th Location Street Address Juneau Park East Mason Street and North Prospect Avenue Geographical Latitude 43° 2'26.48"N Longitude 87°53'56.43"W Coordination Previous Street Address The triangle at North Second Street, North Plankinton Avenue and West Location(s) Wells Street (if applicable) Geographical Latitude 43° 2'25.28"N Longitude 87°54'45.97"W Coordination Dedicated to Revolutionary War Civil War Spanish-American War WWI WWII Vietnam War All wars soldiers of Description Patron(s) or Service Star Legion, Milwaukee Branch Sponsor(s) Designer(s) Benjamin Hawkins (b. 1896): “Artist Benjamin Hawkins received his training from several modem masters of sculptural form- Victor Holm, Leo Lentelli, and Lee Lawrie. Educational experiences at the St. Louis School of Fine Arts, Washington University, the Beaux-Arts Institute of Design, and the Art Students league prepared the artist for a long career. Hawkins moved to New York City as a young man and in 1933 received the Avery Prize from the Architectural league of New York for Boy Riding on a Dolphin. A member of the National Sculpture Society, he chose architectural sculpture as his special field. Hawkins participated in the WPA federal program to decorate post offices during the Great Depression.” (Buck and Palmer, Outdoor Sculpture in Milwaukee, 19-20) Manufacturer(s) N/A Costs $5,000 National State County City Private 1 Current Description Owner(s) Dedication November 11, 1934 Date Other Dates Descriptive Physical Condition Description of The flagpole is located at the southern end of Juneau Park, at N. -

Sculptor Charles Adrian Pillars
CHARLES ADRIAN PILLARS (1870-1937), JACKSONVILLE‟S MOST NOTED SCULPTOR By DIANNE CRUM DAWOOD A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF ARTS UNIVERSITY OF FLORIDA 2011 1 © 2011 Dianne Crum Dawood 2 ACKNOWLEDGMENTS I would first like to acknowledge my thesis chair, Dr. Melissa Hyde, and committee members, Dr. Eric Segal and Dr. Victoria Rovine, for serving on this project for me. Their suggestions, insightful analysis, encouragement, and confidence in my candidacy for a master‟s in art history were important support. I also enjoyed their friendship and patience during this process. The wordsmithing and editing guidance of Mary McClurkin was a delightful collaboration that culminated in a timely finished paper and a treasured friendship. Assistance from Deanne and Ira in the search of microfilm and microfiche records turned a project of anticipated drudgery into a treasure hunt of exciting finds. I also appreciated the suggestions and continued interest of Dr. Wayne Wood, who assured me that Charles Adrian Pillars‟s story was worthy of serious research that culminated in learning details of his life and career heretofore unknown outside of Pillars‟s family. Interviews with Pillars‟s daughter, Ann Pillars Durham, were engaging time travels recalling her father and his celebrity and the family‟s economic and personal vicissitudes during the Great Depression. She also graciously allowed me to review her personal papers. Wells & Drew, the parent company of which was founded in Jacksonville in 1855, permitted my use of a color image, and The Florida Times-Union granted permission to use some of their photographs in this paper.