Hypercalcemia: Pathogenesis, Clinical Manifestations, Differential Diagnosis, and Management
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hyponatremia and Hypernatremia MICHAEL M
This is a corrected version of the article that appeared in print. Diagnosis and Management of Sodium Disorders: Hyponatremia and Hypernatremia MICHAEL M. BRAUN, DO, Madigan Army Medical Center, Tacoma, Washington CRAIG H. BARSTOW, MD, Womack Army Medical Center, Fort Bragg, North Carolina NATASHA J. PYZOCHA, DO, Madigan Army Medical Center, Tacoma, Washington Hyponatremia and hypernatremia are common findings in the inpatient and outpatient settings. Sodium disorders are associated with an increased risk of morbidity and mortality. Plasma osmolality plays a critical role in the patho- physiology and treatment of sodium disorders. Hyponatremia and hypernatremia are classified based on volume status (hypovolemia, euvolemia, and hypervolemia). Sodium disorders are diagnosed by findings from the history, physical examination, laboratory studies, and evaluation of volume status. Treatment is based on symptoms and underlying causes. In general, hyponatremia is treated with fluid restriction (in the setting of euvolemia), isotonic saline (in hypovolemia), and diuresis (in hypervolemia). A combination of these therapies may be needed based on the presentation. Hypertonic saline is used to treat severe symptomatic hyponatremia. Medications such as vaptans may have a role in the treatment of euvolemic and hypervolemic hyponatremia. The treatment of hypernatremia involves correcting the underlying cause and correcting the free water deficit. Am( Fam Physician. 2015;91(5):299-307. Copy- right © 2015 American Academy of Family Physicians.) More online yponatremia is a common elec- a worse prognosis in patients with liver cir- at http://www. trolyte disorder defined as a rhosis, pulmonary hypertension, myocardial aafp.org/afp. serum sodium level of less than infarction, chronic kidney disease, hip frac- CME This clinical content 135 mEq per L.1-3 A Dutch sys- tures, and pulmonary embolism.1,8-10 conforms to AAFP criteria Htematic review of 53 studies showed that the for continuing medical Etiology and Pathophysiology education (CME). -

Dysmagnesemia in Covid-19 Cohort Patients: Prevalence and Associated Factors
Magnesium Research 2020; 33 (4): 114-122 ORIGINAL ARTICLE Dysmagnesemia in Covid-19 cohort patients: prevalence and associated factors Didier Quilliot1, Olivier Bonsack1, Roland Jaussaud2, Andre´ Mazur3 1 Transversal Nutrition Unit and; 2 Internal Medicine and Clinical Immunology. Nancy University Hospital, University of Lorraine, France; 3 Universite´ Clermont Auvergne, INRAE, UNH, Unite´ de Nutrition Humaine, Clermont-Ferrand, France Correspondence <[email protected]> Abstract. Hypomagnesemia and hypermagnesemia could have serious implications and possibly lead to progress from a mild form to a severe outcome of Covid-19. Susceptibility of subjects with low magnesium status to develop and enhance this infection is possible. There is little data on the magnesium status of patients with Covid-19 with different degrees of severity. This study was conducted to evaluate prevalence of dysmagnesemia in a prospective Covid-19 cohort study according to the severity of the clinical manifestations and to identify factors associated. Serum magnesium was measured in 300 of 549 patients admitted to the hospital due to severe Covid-19. According to the WHO guidelines, patients were classified as moderate, severe, or critical. 48% patients had a magnesemia below 0.75 mmol/L (defined as magnesium deficiency) including 13% with a marked hypomagnesemia (<0.65 mmol/L). 9.6% had values equal to or higher than 0.95 mmol/L. Serum magnesium concentrations were significantly lower in female than in male (0.73 Æ 0.12 vs 0.80 Æ 0.13 mmol/L), whereas the sex ratio M/F was higher in severe and critical form (p<0.001). In a bivariate analysis, the risk of magnesium deficiency was significantly and negatively associated with infection severity (p<0.001), sex ratio (M/F, p<0.001), oxygenotherapy (p<0.001), stay in critical care unit (p=0.028), and positively with nephropathy (p=0.026). -

Drugs for the Treatment of Hypercalcaemia C. R. PATERSON
Postgrad Med J: first published as 10.1136/pgmj.50.581.158 on 1 March 1974. Downloaded from Postgraduate Medical Journal (March 1974) 50, 158-162. Drugs for the treatment of hypercalcaemia C. R. PATERSON M.A., D.M., M.R.C.PATH. Department of Clinical Chemistry, University of Dundee, Dundee Summary able commercially (Phosphate Sandoz). The initial Hypercalcaemia is ordinarily treated by treatment of dose is 1-3 g (of phosphorus) per day according to the underlying disorder. In some cases, as in malignant the size of the patient. The serum calcium should be disease, in vitamin D poisoning and after a failed checked daily at first and the dose adjusted appro- parathyroidectomy, the hypercalcaemia itself needs to priately so that the maintenace dose is the minimum be treated. A large number of methods have been consistent with control of the serum calcium. Oral advocated for this, but phosphate is the drug of choice phosphate therapy is effective in the management of in most patients. This paper outlines its use, mode of hypercalcaemia of malignant disease and myeloma action and side effects and reviews the other methods (Goldsmith and Ingbar, 1966; Goldsmith et al., proposed for the management of hypercalcaemia. 1968; Thalassinos and Joplin, 1968), in hyperpara- thyroidism (Dent, 1962; Goldsmith and Ingbar, 1966; Eisenberg, 1968), and in vitamin D poisoningProtected by copyright. Introduction (Goldsmith and Ingbar, 1966). The management of hypercalcaemia is ordinarily Intravenous phosphate therapy is used in the that of the underlying disorder (Wills, 1971; management of acute hypercalcaemia, especially in Henrard, 1971; Watson, 1972). There are, however, the patient who is vomiting or in coma. -

Epileptic Seizure, As the First Symptom of Hypoparathyroidism in Children, Does Not Require Antiepileptic Drugs
Childs Nerv Syst DOI 10.1007/s00381-016-3264-2 ORIGINAL PAPER Epileptic seizure, as the first symptom of hypoparathyroidism in children, does not require antiepileptic drugs Meng-Jia Liu1 & Jiu-Wei Li2 & Xiu-Yu Shi1 & Lin-Yan Hu1 & Li-Ping Zou1,3 Received: 28 May 2016 /Accepted: 3 October 2016 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Introduction Objective Patients with hypoparathyroidism exhibit metabol- ic disorders (hypocalcemia) and brain structural abnormalities Epileptic seizure occurs when a burst of electrical impulses in (brain calcifications). Currently, studies have determined the brain exceeds the normal limits. Its manifestation can vary whether antiepileptic drug (AED) treatment is required for from uncontrolled jerking movement (tonic–clonic seizure) to epileptic seizures in children with hypoparathyroidism. momentary loss of awareness (absence seizure). These im- Method This study aims to evaluate the data of two medical pulses spread to adjacent areas in the brain and create an un- centers in Beijing based on the diagnosis of epileptic seizures controlled storm of electrical activity. Brain diseases character- as the first symptom of hypoparathyroidism in children. ized by enduring predisposition to generate epileptic seizures Result A total of 42 patients were included and assigned into are collectively called epilepsy. According to pathogenesis, ep- AED and non-AED treatment groups in a 1:2 matched case– ilepsy can be classified into six categories: metabolic, structural, control study. Results show that the seizure outcome after inherited, immunologic, inflammatory, and idiopathic. 1 year of AED treatment is not significantly different from Hypoparathyroidism is an endocrine disease that results that of the control. -

A Case of Calciphylaxis with an Unfavorable Outcome
CASE LETTER 671 However, the present patient exhibited the peculiarity References of an intense inflammatory infiltrate in the dermis rarely seen previously. According to this case, an atypical intense 1. Sitaru C, Zillikens D. Mechanisms of blister induction by autoan- inflammation could be a distinct histopathological feature tibodies. Exp Dermatol. 2005;14:861---75. of trauma-induced pemphigus. Further research is neces- 2. Sagi L, Trau H. The Koebner phenomenon. Clin Dermatol. sary to ascertain if greater inflammation in dermis is more 2011;29:231---6. related to trauma-induced pemphigus as opposed to those 3. Jang HW, Chun SH, Lee JM, Jeon J, Hashimoto T, Kim IH. Radiotherapy-induced pemphigus vulgaris. J Dermatol. not related to trauma. 2014;41:851---2. In conclusion, this report describes a presumably new 4. Daneshpazhooh M, Fatehnejad M, Rahbar Z, Balighi K, case of trauma-induced pemphigus. To the author’s knowl- Ghandi N, Ghiasi M, et al. Trauma-induced pemphigus: a edge, there are no prior reports of pemphigus involving case series of 36 patients. J Dtsch Dermatol Ges. 2016;14: predominantly the breasts with such a peculiar dispo- 166---71. sition. It is proposed that a hypothetical continuous 5. Balighi K, Daneshpazhooh M, Azizpour A, Lajevardi V, trauma with the bra could explain this unique loca- Mohammadi F, Chams-Davatchi C. Koebner phenomenon tion. in pemphigus vulgaris patients. JAAD Case Rep. 2016;2: 419---21. Financial support Fernando Garcia-Souto None declared. Department of Dermatology, Valme University Hospital, Seville, Spain Author’s contributions E-mail: [email protected] Received 14 November 2019; accepted 5 March 2020 Fernando Garcia-Souto: Conception and planning of the manuscript; writing and critical revision of the manuscript; https://doi.org/10.1016/j.abd.2020.03.010 approval of the final version. -

Extensive Skin Necrosis Ulcers and Systemic Vascular Insufficiency Syndrome with Calciphylaxis: a Case Report
Urology & Nephrology Open Access Journal Case Report Open Access Extensive skin necrosis ulcers and systemic vascular insufficiency syndrome with calciphylaxis: a case report Abstract Volume 5 Issue 4 - 2017 Calciphylaxis is a part of systemic vascular calcification that is commonly seen in patients 1 2,3 with end stage renal disease. It presents as skin ischemia and necrosis due to calcification Alexander M Swan, Nancy J Fried, 2,3 2 of dermal arterioles. There is no consensus on optimal treatment and the condition is Charisma Lee, Thein Aung, Zaw Nyein associated with a high mortality rate. Here we report a patient on peritoneal dialysis with Aye,2 Deepthi Gunasekaran2 a high calcium-phosphorus product and extensive calciphylaxis involving the extremities, 1Wilkes University, USA back and scalp. We used a multi-interventional approach to treat this patient with a good 2Nephrology Hypertension Renal transplant and Renal Therapy, outcome. The underlying pathology of systemic vascular calcification and insufficiency USA 3 may involve other vascular beds such as the coronary and cerebral vasculature. Early Rutgers New Jersey Medical School, USA detection of these conditions may improve outcomes in these patents. Correspondence: Alexander M Swan, AA Prof of Medicine, Keywords: necrotic ulcer of skin, calciphylaxis, calcium-phosphorus product, vascular Rutgers New Jersey Medical School, 185 S Orange Ave, Newark, calcification, systemic vascular insufficiency NJ 07103, President, Garden State Kidney Center, 345 Main Street, Woodbridge, NJ 07095, USA, Tel 732-750-5555, Fax 732- 750-5550, Email Received: November 10, 2017 | Published: October 26, 2017 Abbreviations: ESRD, end-stage renal disease; CUA, calcific secondary to hypertension and peritoneal dialys is was initiated 5years uremic arterilopathy; LMWH, low molecular weight heparin ago after multiple access problems with hemodialysis. -

A Case of Cushing Syndrome with Both Secondary Hypothyroidism and Hypercalcemia Due to Postoperative Adrenal Insufficiency
Endocrine Journal 2004, 51 (1), 105–113 NOTE A Case of Cushing Syndrome with Both Secondary Hypothyroidism and Hypercalcemia Due to Postoperative Adrenal Insufficiency MASAHITO KATAHIRA, TSUTOMU YAMADA* AND MASAHIKO KAWAI* Department of Internal Medicine, Kyoritsu General Hospital, Nagoya 456-8611, Japan *Division of Endocrinology, Department of Internal Medicine, Okazaki City Hospital, Okazaki 444-8553, Japan Abstract. A 48-year-old woman was referred to our hospital because of secondary hypothyroidism. Upon admission a left adrenal tumor was also detected using computed tomography. Laboratory data and adrenal scintigraphy were compatible with Cushing syndrome due to the left adrenocortical adenoma, although she showed no response to the TRH stimulation test. Hypercortisolism resulting in secondary hypothyroidism was diagnosed. After a left adrenalectomy, hydrocortisone administration was begun and the dose was reduced gradually. After discharge on the 23rd postoperative day, she began to suffer from anorexia. ACTH level remained low, and serum cortisol, free thyroxine and TSH levels were within the normal range. Since her condition became worse, she was re-admitted on the 107th postoperative day at which time serum calcium level was high (15.6 mg/dl). Both ACTH response to the CRH stimulation test and TSH response to the TRH stimulation test were restored to almost normal levels, but there was no response of cortisol to CRH stimulation test. We diagnosed that the hypercalcemia was due to adrenal insufficiency. Although the serum calcium level decreased to normal after hydrocortisone was increased (35 mg/day), secondary hypothyroidism recurred. It was suggested that sufficient glucocorticoids suppressed TSH secretion mainly at the pituitary level, which resulted in secondary (corticogenic) hypothyroidism. -
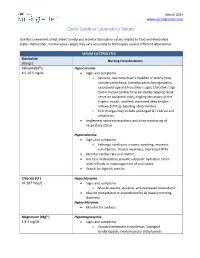
Quick Guide to Laboratory Values
March 2021 www.nursingcenter.com Quick Guide to Laboratory Values Use this convenient cheat-sheet to help you monitor laboratory values related to fluid and electrolyte status. Remember, normal value ranges may vary according to techniques used in different laboratories. SERUM ELECTROLYTES Electrolyte Nursing Considerations (Range) Calcium (Ca2+) Hypocalcemia 8.5-10.5 mg/dL • Signs and symptoms o Seizures, neuromuscular irritability or tetany (may include paresthesia, bronchospasm, laryngospasm, carpopedal spasm [Trousseau’s sign], Chvostek’s sign [facial muscle contractions elicited by tapping facial nerve on ipsilateral side], tingling sensations of the fingers, mouth, and feet, increased deep tendon reflexes [DTRs]), bleeding abnormalities o ECG changes may include prolonged QT interval and arrythmias. • Implement seizure precautions and close monitoring of respiratory status. Hypercalcemia • Signs and symptoms o Lethargy, confusion, nausea, vomiting, anorexia, constipation, muscle weakness, depressed DTRs • Monitor cardiac rate and rhythm. • Increase mobilization, provide adequate hydration either with IV fluids or encouragement of oral intake. • Watch for digitalis toxicity. Chloride (Cl-) Hypochloremia 97-107 mEq/L • Signs and symptoms o Muscle spasms, alkalosis, and depressed respirations • May be precipitated or exacerbated by GI losses (vomiting, diarrhea). Hyperchloremia • Monitor for acidosis. Magnesium (Mg2+) Hypomagnesemia 1.8-3 mg/dL • Signs and symptoms o Cardiac/ventricular arrhythmias, laryngeal stridor/spasm, neuromuscular -

PARANEOPLASTIC SYNDROMES: J Neurol Neurosurg Psychiatry: First Published As 10.1136/Jnnp.2004.040378 on 14 May 2004
PARANEOPLASTIC SYNDROMES: J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.2004.040378 on 14 May 2004. Downloaded from WHEN TO SUSPECT, HOW TO CONFIRM, AND HOW TO MANAGE ii43 J H Rees J Neurol Neurosurg Psychiatry 2004;75(Suppl II):ii43–ii50. doi: 10.1136/jnnp.2004.040378 eurological manifestations of cancer are common, disabling, and often multifactorial (table 1). The concept that malignant disease can cause damage to the nervous system Nabove and beyond that caused by direct or metastatic infiltration is familiar to all clinicians looking after cancer patients. These ‘‘remote effects’’ or paraneoplastic manifestations of cancer include metabolic and endocrine syndromes such as hypercalcaemia, and the syndrome of inappropriate ADH (antidiuretic hormone) secretion. Paraneoplastic neurological disorders (PNDs) are remote effects of systemic malignancies that affect the nervous system. The term PND is reserved for those disorders that are caused by an autoimmune response directed against antigens common to the tumour and nerve cells. PNDs are much less common than direct, metastatic, and treatment related complications of cancer, but are nevertheless important because they cause severe neurological morbidity and mortality and frequently present to the neurologist in a patient without a known malignancy. Because of the relative rarity of PND, neurological dysfunction should only be regarded as paraneoplastic if a particular neoplasm associates with a remote but specific effect on the nervous system more frequently than would be expected by chance. For example, subacute cerebellar ataxia in the setting of ovarian cancer is sufficiently characteristic to be called paraneoplastic cerebellar degeneration, as long as other causes have been ruled out. -

Magnesium: the Forgotten Electrolyte—A Review on Hypomagnesemia
medical sciences Review Magnesium: The Forgotten Electrolyte—A Review on Hypomagnesemia Faheemuddin Ahmed 1,* and Abdul Mohammed 2 1 OSF Saint Anthony Medical Center, 5666 E State St, Rockford, IL 61108, USA 2 Advocate Illinois Masonic Medical Center, 833 W Wellington Ave, Chicago, IL 60657, USA; [email protected] * Correspondence: [email protected] Received: 20 February 2019; Accepted: 2 April 2019; Published: 4 April 2019 Abstract: Magnesium is the fourth most abundant cation in the body and the second most abundant intracellular cation. It plays an important role in different organ systems at the cellular and enzymatic levels. Despite its importance, it still has not received the needed attention either in the medical literature or in clinical practice in comparison to other electrolytes like sodium, potassium, and calcium. Hypomagnesemia can lead to many clinical manifestations with some being life-threatening. The reported incidence is less likely than expected in the general population. We present a comprehensive review of different aspects of magnesium physiology and hypomagnesemia which can help clinicians in understanding, identifying, and treating this disorder. Keywords: magnesium; proton pump inhibitors; diuretics; hypomagnesemia 1. Introduction Magnesium is one of the most abundant cation in the body as well as an abundant intracellular cation. It plays an important role in molecular, biochemical, physiological, and pharmacological functions in the body. The importance of magnesium is well known, but still it is the forgotten electrolyte. The reason for it not getting the needed attention is because of rare symptomatology until levels are really low and also because of a lack of proper understanding of magnesium physiology. -

AACE Annual Meeting 2021 Abstracts Editorial Board
June 2021 Volume 27, Number 6S AACE Annual Meeting 2021 Abstracts Editorial board Editor-in-Chief Pauline M. Camacho, MD, FACE Suleiman Mustafa-Kutana, BSC, MB, CHB, MSC Maywood, Illinois, United States Boston, Massachusetts, United States Vin Tangpricha, MD, PhD, FACE Atlanta, Georgia, United States Andrea Coviello, MD, MSE, MMCi Karel Pacak, MD, PhD, DSc Durham, North Carolina, United States Bethesda, Maryland, United States Associate Editors Natalie E. Cusano, MD, MS Amanda Powell, MD Maria Papaleontiou, MD New York, New York, United States Boston, Massachusetts, United States Ann Arbor, Michigan, United States Tobias Else, MD Gregory Randolph, MD Melissa Putman, MD Ann Arbor, Michigan, United States Boston, Massachusetts, United States Boston, Massachusetts, United States Vahab Fatourechi, MD Daniel J. Rubin, MD, MSc Harold Rosen, MD Rochester, Minnesota, United States Philadelphia, Pennsylvania, United States Boston, Massachusetts, United States Ruth Freeman, MD Joshua D. Safer, MD Nicholas Tritos, MD, DS, FACP, FACE New York, New York, United States New York, New York, United States Boston, Massachusetts, United States Rajesh K. Garg, MD Pankaj Shah, MD Boston, Massachusetts, United States Staff Rochester, Minnesota, United States Eliza B. Geer, MD Joseph L. Shaker, MD Paul A. Markowski New York, New York, United States Milwaukee, Wisconsin, United States CEO Roma Gianchandani, MD Lance Sloan, MD, MS Elizabeth Lepkowski Ann Arbor, Michigan, United States Lufkin, Texas, United States Chief Learning Officer Martin M. Grajower, MD, FACP, FACE Takara L. Stanley, MD Lori Clawges The Bronx, New York, United States Boston, Massachusetts, United States Senior Managing Editor Allen S. Ho, MD Devin Steenkamp, MD Corrie Williams Los Angeles, California, United States Boston, Massachusetts, United States Peer Review Manager Michael F. -

Increased Mortality Associated with Hypermagnesemia in Severe COVID-19 Illness
Original Investigation Increased Mortality Associated with Hypermagnesemia in Severe COVID-19 Illness Jacob S. Stevens 1,2, Andrew A. Moses1, Thomas L. Nickolas1,2, Syed Ali Husain1,2, and Sumit Mohan1,2,3 Key Points Hypermagnesemia is common in patients admitted with coronavirus disease 2019. The development of hypermagnesemia in coronavirus disease 2019 is associated with renal failure and markers of high cell turnover. In adjusted models, patients who develop hypermagnesemia have an increased risk of mortality. Abstract Background Although electrolyte abnormalities are common among patients with COVID-19, very little has been reported on magnesium homeostasis in these patients. Here we report the incidence of hypermagnesemia, and its association with outcomes among patients admitted with COVID-19. Methods We retrospectively identified all patients with a positive test result for SARS-CoV-2who were admitted to a large quaternary care center in New York City in spring 2020. Details of the patients’ demographics and hospital course were obtained retrospectively from medical records. Patients were defined as having hypermagnesemia if their median magnesium over the course of their hospitalization was .2.4 mg/dl. Results A total of 1685 patients hospitalized with COVID-19 had their magnesium levels checked during their hospitalization, and were included in the final study cohort, among whom 355 (21%) had hypermagnesemia. Patients who were hypermagnesemic had a higher incidence of shock requiring pressors (35% vs 27%, P,0.01), respiratory failure requiring mechanical ventilation (28% vs 21%, P50.01), AKI (65% vs 50%, P,0.001), and AKI severe enough to require renal replacement therapy (18% vs 5%, P,0.001).