Proteomic and Transcriptomic Profiling Reveal Different Aspects of Aging in the Kidney
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
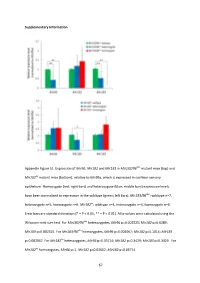
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

KCC4/SLC12A7 Polyclonal Antibody Catalog Number:26590-1-AP
For Research Use Only KCC4/SLC12A7 Polyclonal antibody www.ptglab.com Catalog Number:26590-1-AP Catalog Number: GenBank Accession Number: Purification Method: Basic Information 26590-1-AP BC098390 Antigen affinity purification Size: GeneID (NCBI): Recommended Dilutions: 150ul , Concentration: 900 μg/ml by 10723 WB 1:2000-1:10000 Nanodrop; Full Name: Source: solute carrier family 12 Rabbit (potassium/chloride transporters), Isotype: member 7 IgG Immunogen Catalog Number: AG24954 Applications Tested Applications: Positive Controls: WB, ELISA WB : BGC-823 cells, NCI-H1299 cells, Raji cells, SH- Species Specificity: SY5Y cells human KCC4, also named as Solute carrier family 12 member 7(SLC12A7), is a 1083 amino-acid protein, which belongs to Background Information the SLC12A transporter family. As a multi-pass membrane protein, the molecular weight of KCC4 is 119 kDa. KCC4 mediates electroneutral potassium-chloride cotransport when activated by cell swelling. Moreover, KCC4 may mediate K+ uptake into Deiters' cells in the cochlea and contribute to K+ recycling in the inner ear. KCC4 is important for the survival of cochlear outer and inner hair cells and the maintenance of the organ of Corti(Uniprot, GeneID:10723). Storage: Storage Store at -20°C. Stable for one year after shipment. Storage Buffer: PBS with 0.02% sodium azide and 50% glycerol pH 7.3. Aliquoting is unnecessary for -20ºC storage For technical support and original validation data for this product please contact: This product is exclusively available under Proteintech T: 1 (888) 4PTGLAB (1-888-478-4522) (toll free E: [email protected] Group brand and is not available to purchase from any in USA), or 1(312) 455-8498 (outside USA) W: ptglab.com other manufacturer. -

The Genetic Landscape of the Human Solute Carrier (SLC) Transporter Superfamily
Human Genetics (2019) 138:1359–1377 https://doi.org/10.1007/s00439-019-02081-x ORIGINAL INVESTIGATION The genetic landscape of the human solute carrier (SLC) transporter superfamily Lena Schaller1 · Volker M. Lauschke1 Received: 4 August 2019 / Accepted: 26 October 2019 / Published online: 2 November 2019 © The Author(s) 2019 Abstract The human solute carrier (SLC) superfamily of transporters is comprised of over 400 membrane-bound proteins, and plays essential roles in a multitude of physiological and pharmacological processes. In addition, perturbation of SLC transporter function underlies numerous human diseases, which renders SLC transporters attractive drug targets. Common genetic polymorphisms in SLC genes have been associated with inter-individual diferences in drug efcacy and toxicity. However, despite their tremendous clinical relevance, epidemiological data of these variants are mostly derived from heterogeneous cohorts of small sample size and the genetic SLC landscape beyond these common variants has not been comprehensively assessed. In this study, we analyzed Next-Generation Sequencing data from 141,456 individuals from seven major human populations to evaluate genetic variability, its functional consequences, and ethnogeographic patterns across the entire SLC superfamily of transporters. Importantly, of the 204,287 exonic single-nucleotide variants (SNVs) which we identifed, 99.8% were present in less than 1% of analyzed alleles. Comprehensive computational analyses using 13 partially orthogonal algorithms that predict the functional impact of genetic variations based on sequence information, evolutionary conserva- tion, structural considerations, and functional genomics data revealed that each individual genome harbors 29.7 variants with putative functional efects, of which rare variants account for 18%. Inter-ethnic variability was found to be extensive, and 83% of deleterious SLC variants were only identifed in a single population. -

Nephritis Responses in Murine and Human Lupus Analysis Defines
Downloaded from http://www.jimmunol.org/ by guest on October 2, 2021 is online at: average * The Journal of Immunology , 24 of which you can access for free at: 2012; 189:988-1001; Prepublished online 20 June from submission to initial decision 4 weeks from acceptance to publication Celine C. Berthier, Ramalingam Bethunaickan, Tania Gonzalez-Rivera, Viji Nair, Meera Ramanujam, Weijia Zhang, Erwin P. Bottinger, Stephan Segerer, Maja Lindenmeyer, Clemens D. Cohen, Anne Davidson and Matthias Kretzler 2012; doi: 10.4049/jimmunol.1103031 http://www.jimmunol.org/content/189/2/988 Cross-Species Transcriptional Network Analysis Defines Shared Inflammatory Responses in Murine and Human Lupus Nephritis J Immunol cites 60 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://www.jimmunol.org/content/189/2/988.full#ref-list-1 http://www.jimmunol.org/content/suppl/2012/06/20/jimmunol.110303 1.DC1 This article Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of October 2, 2021. The Journal of Immunology Cross-Species Transcriptional Network Analysis Defines Shared Inflammatory Responses in Murine and Human Lupus Nephritis Celine C. -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

SLC12A6 Gene Solute Carrier Family 12 Member 6
SLC12A6 gene solute carrier family 12 member 6 Normal Function The SLC12A6 gene provides instructions for making a protein called a K-Cl cotransporter. This protein is involved in moving charged atoms (ions) of potassium (K) and chlorine (Cl) across the cell membrane. The positively charged potassium ions and negatively charged chlorine ions are moved together (cotransported), so that the charges inside and outside the cell membrane are unchanged (electroneutral). Electroneutral cotransport of ions across cell membranes is involved in many functions of the body. While the specific function of the K-Cl cotransporter produced from the SLC12A6 gene is unknown, it seems to be critical for the development and maintenance of nerve tissue. It may be involved in regulating the amounts of potassium, chlorine, or water in cells and intercellular spaces. The K-Cl cotransporter protein may also help regulate the activity of other proteins that are sensitive to ion concentrations. Health Conditions Related to Genetic Changes Andermann syndrome At least six SLC12A6 gene mutations have been identified in people with Andermann syndrome. Almost all affected individuals of French-Canadian descent have the same mutation in both copies of the SLC12A6 gene, in which the DNA building block ( nucleotide) guanine is deleted at position 2436 (written as 2436delG). This mutation is common in the populations of the Saguenay-Lac-St.-Jean and Charlevoix regions of northeastern Quebec. Most SLC12A6 gene mutations that cause Andermann syndrome result in a K-Cl cotransporter protein that is shortened and nonfunctional. The lack of functional protein produced from the SLC12A6 gene is believed to interfere with the development of the corpus callosum and maintenance of the nerves that transmit signals needed for movement and sensation, resulting in the signs and symptoms of Andermann syndrome. -

MAP17's Up-Regulation, a Crosspoint in Cancer and Inflammatory
García-Heredia and Carnero Molecular Cancer (2018) 17:80 https://doi.org/10.1186/s12943-018-0828-7 REVIEW Open Access Dr. Jekyll and Mr. Hyde: MAP17’s up- regulation, a crosspoint in cancer and inflammatory diseases José M. García-Heredia1,2,3 and Amancio Carnero1,3* Abstract Inflammation is a common defensive response that is activated after different harmful stimuli. This chronic, or pathological, inflammation is also one of the causes of neoplastic transformation and cancer development. MAP17 is a small protein localized to membranes with a restricted pattern of expression in adult tissues. However, its expression is common in destabilized cells, as it is overexpressed both in inflammatory diseases and in cancer. MAP17 is overexpressed in most, if not all, carcinomas and in many tumors of mesenchymal origin, and correlates with higher grade and poorly differentiated tumors. This overexpression drives deep changes in cell homeostasis including increased oxidative stress, deregulation of signaling pathways and increased growth rates. Importantly, MAP17 is associated in tumors with inflammatory cells infiltration, not only in cancer but in various inflammatory diseases such as Barret’s esophagus, lupus, Crohn’s, psoriasis and COPD. Furthermore, MAP17 also modifies the expression of genes connected to inflammation, showing a clear induction of the inflammatory profile. Since MAP17 appears highly correlated with the infiltration of inflammatory cells in cancer, is MAP17 overexpression an important cellular event connecting tumorigenesis and inflammation? Keywords: MAP17, Cancer, Inflammatory diseases Background process ends when the activated cells undergo apoptosis in Inflammatory response is a common defensive process acti- a highly regulated process that finishes after pathogens and vated after different harmful stimuli, constituting a highly cell debris have been phagocytized [9]. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Genetic Background of Ataxia in Children Younger Than 5 Years in Finland E444
Volume 6, Number 4, August 2020 Neurology.org/NG A peer-reviewed clinical and translational neurology open access journal ARTICLE Genetic background of ataxia in children younger than 5 years in Finland e444 ARTICLE Cerebral arteriopathy associated with heterozygous variants in the casitas B-lineage lymphoma gene e448 ARTICLE Somatic SLC35A2 mosaicism correlates with clinical fi ndings in epilepsy brain tissuee460 ARTICLE Synonymous variants associated with Alzheimer disease in multiplex families e450 Academy Officers Neurology® is a registered trademark of the American Academy of Neurology (registration valid in the United States). James C. Stevens, MD, FAAN, President Neurology® Genetics (eISSN 2376-7839) is an open access journal published Orly Avitzur, MD, MBA, FAAN, President Elect online for the American Academy of Neurology, 201 Chicago Avenue, Ann H. Tilton, MD, FAAN, Vice President Minneapolis, MN 55415, by Wolters Kluwer Health, Inc. at 14700 Citicorp Drive, Bldg. 3, Hagerstown, MD 21742. Business offices are located at Two Carlayne E. Jackson, MD, FAAN, Secretary Commerce Square, 2001 Market Street, Philadelphia, PA 19103. Production offices are located at 351 West Camden Street, Baltimore, MD 21201-2436. Janis M. Miyasaki, MD, MEd, FRCPC, FAAN, Treasurer © 2020 American Academy of Neurology. Ralph L. Sacco, MD, MS, FAAN, Past President Neurology® Genetics is an official journal of the American Academy of Neurology. Journal website: Neurology.org/ng, AAN website: AAN.com CEO, American Academy of Neurology Copyright and Permission Information: Please go to the journal website (www.neurology.org/ng) and click the Permissions tab for the relevant Mary E. Post, MBA, CAE article. Alternatively, send an email to [email protected]. -

Strand Breaks for P53 Exon 6 and 8 Among Different Time Course of Folate Depletion Or Repletion in the Rectosigmoid Mucosa
SUPPLEMENTAL FIGURE COLON p53 EXONIC STRAND BREAKS DURING FOLATE DEPLETION-REPLETION INTERVENTION Supplemental Figure Legend Strand breaks for p53 exon 6 and 8 among different time course of folate depletion or repletion in the rectosigmoid mucosa. The input of DNA was controlled by GAPDH. The data is shown as ΔCt after normalized to GAPDH. The higher ΔCt the more strand breaks. The P value is shown in the figure. SUPPLEMENT S1 Genes that were significantly UPREGULATED after folate intervention (by unadjusted paired t-test), list is sorted by P value Gene Symbol Nucleotide P VALUE Description OLFM4 NM_006418 0.0000 Homo sapiens differentially expressed in hematopoietic lineages (GW112) mRNA. FMR1NB NM_152578 0.0000 Homo sapiens hypothetical protein FLJ25736 (FLJ25736) mRNA. IFI6 NM_002038 0.0001 Homo sapiens interferon alpha-inducible protein (clone IFI-6-16) (G1P3) transcript variant 1 mRNA. Homo sapiens UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 15 GALNTL5 NM_145292 0.0001 (GALNT15) mRNA. STIM2 NM_020860 0.0001 Homo sapiens stromal interaction molecule 2 (STIM2) mRNA. ZNF645 NM_152577 0.0002 Homo sapiens hypothetical protein FLJ25735 (FLJ25735) mRNA. ATP12A NM_001676 0.0002 Homo sapiens ATPase H+/K+ transporting nongastric alpha polypeptide (ATP12A) mRNA. U1SNRNPBP NM_007020 0.0003 Homo sapiens U1-snRNP binding protein homolog (U1SNRNPBP) transcript variant 1 mRNA. RNF125 NM_017831 0.0004 Homo sapiens ring finger protein 125 (RNF125) mRNA. FMNL1 NM_005892 0.0004 Homo sapiens formin-like (FMNL) mRNA. ISG15 NM_005101 0.0005 Homo sapiens interferon alpha-inducible protein (clone IFI-15K) (G1P2) mRNA. SLC6A14 NM_007231 0.0005 Homo sapiens solute carrier family 6 (neurotransmitter transporter) member 14 (SLC6A14) mRNA. -

Epigenetic Mechanisms Are Involved in the Oncogenic Properties of ZNF518B in Colorectal Cancer
Epigenetic mechanisms are involved in the oncogenic properties of ZNF518B in colorectal cancer Francisco Gimeno-Valiente, Ángela L. Riffo-Campos, Luis Torres, Noelia Tarazona, Valentina Gambardella, Andrés Cervantes, Gerardo López-Rodas, Luis Franco and Josefa Castillo SUPPLEMENTARY METHODS 1. Selection of genomic sequences for ChIP analysis To select the sequences for ChIP analysis in the five putative target genes, namely, PADI3, ZDHHC2, RGS4, EFNA5 and KAT2B, the genomic region corresponding to the gene was downloaded from Ensembl. Then, zoom was applied to see in detail the promoter, enhancers and regulatory sequences. The details for HCT116 cells were then recovered and the target sequences for factor binding examined. Obviously, there are not data for ZNF518B, but special attention was paid to the target sequences of other zinc-finger containing factors. Finally, the regions that may putatively bind ZNF518B were selected and primers defining amplicons spanning such sequences were searched out. Supplementary Figure S3 gives the location of the amplicons used in each gene. 2. Obtaining the raw data and generating the BAM files for in silico analysis of the effects of EHMT2 and EZH2 silencing The data of siEZH2 (SRR6384524), siG9a (SRR6384526) and siNon-target (SRR6384521) in HCT116 cell line, were downloaded from SRA (Bioproject PRJNA422822, https://www.ncbi. nlm.nih.gov/bioproject/), using SRA-tolkit (https://ncbi.github.io/sra-tools/). All data correspond to RNAseq single end. doBasics = TRUE doAll = FALSE $ fastq-dump -I --split-files SRR6384524 Data quality was checked using the software fastqc (https://www.bioinformatics.babraham. ac.uk /projects/fastqc/). The first low quality removing nucleotides were removed using FASTX- Toolkit (http://hannonlab.cshl.edu/fastxtoolkit/).