The Genetic Architecture of Fasting Plasma Triglyceride Response to Fenofibrate Treatment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Expression of the Human Apolipoprotein Genes and Their Regulation by Ppars
CORE Metadata, citation and similar papers at core.ac.uk Provided by UEF Electronic Publications The expression of the human apolipoprotein genes and their regulation by PPARs Juuso Uski M.Sc. Thesis Biochemistry Department of Biosciences University of Kuopio June 2008 Abstract The expression of the human apolipoprotein genes and their regulation by PPARs. UNIVERSITY OF KUOPIO, the Faculty of Natural and Environmental Sciences, Curriculum of Biochemistry USKI Juuso Oskari Thesis for Master of Science degree Supervisors Prof. Carsten Carlberg, Ph.D. Merja Heinäniemi, Ph.D. June 2008 Keywords: nuclear receptors; peroxisome proliferator-activated receptor; PPAR response element; apolipoprotein; lipid metabolism; high density lipoprotein; low density lipoprotein. Lipids are any fat-soluble, naturally-occurring molecules and one of their main biological functions is energy storage. Lipoproteins carry hydrophobic lipids in the water and salt-based blood environment for processing and energy supply in liver and other organs. In this study, the genomic area around the apolipoprotein genes was scanned in silico for PPAR response elements (PPREs) using the in vitro data-based computer program. Several new putative REs were found in surroundings of multiple lipoprotein genes. The responsiveness of those apolipoprotein genes to the PPAR ligands GW501516, rosiglitazone and GW7647 in the HepG2, HEK293 and THP-1 cell lines were tested with real-time PCR. The APOA1, APOA2, APOB, APOD, APOE, APOF, APOL1, APOL3, APOL5 and APOL6 genes were found to be regulated by PPARs in direct or secondary manners. Those results provide new insights in the understanding of lipid metabolism and so many lifestyle diseases like atherosclerosis, type 2 diabetes, heart disease and stroke. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Proteomic Analysis of HDL from Inbred Mouse Strains Implicates APOE Associated with HDL in Reduced Cholesterol Effl Ux Capacity Via the ABCA1 Pathway
Supplemental Material can be found at: http://www.jlr.org/content/suppl/2015/12/15/jlr.M063701.DC1 .html ˔ Author’s Choice Proteomic analysis of HDL from inbred mouse strains implicates APOE associated with HDL in reduced cholesterol effl ux capacity via the ABCA1 pathway Nathalie Pamir , 1, * Patrick Hutchins , * Graziella Ronsein , * Tomas Vaisar , * Catherine A. Reardon , † Godfrey S. Getz , † Aldons J. Lusis , § and Jay W. Heinecke * Downloaded from Department of Medicine,* University of Washington , Seattle, WA ; Department of Pathology, † University of Chicago , Chicago, IL ; and Department of Genetics, § University of California at Los Angeles , Los Angeles, CA Abstract Cholesterol effl ux capacity associates strongly Supplementary key words atherosclerosis • cardiovascular risk • mass and negatively with the incidence and prevalence of human spectrometry • high density lipoprotein • apolipoprotein E • ATP bind- CVD. We investigated the relationships of HDL’s size and ing cassette transporter A1 www.jlr.org protein cargo with its cholesterol effl ux capacity using APOB-depleted serum and HDLs isolated from fi ve inbred mouse strains with different susceptibilities to atherosclero- Clinical and epidemiological studies show a robust at Univ of Washington Health Sciences Library SB-55, on February 5, 2016 sis. Like humans, mouse HDL carried >70 proteins linked to inverse association of HDL-cholesterol (HDL-C) levels lipid metabolism, the acute-phase response, proteinase inhi- with CVD risk ( 1 ). In randomized clinical trials, how- bition, and the immune system. HDL’s content of specifi c ever, two drugs, CETP inhibitors and niacin, that elevate proteins strongly correlated with its size and cholesterol ef- fl ux capacity, suggesting that its protein cargo regulates its HDL-C levels by different mechanisms, failed to reduce function. -

Biomolecules
biomolecules Review High-Density Lipoproteins Are Bug Scavengers Olivier Meilhac 1,2,*, Sébastien Tanaka 1,3 and David Couret 1,4 1 Université de la Réunion, Inserm, UMR 1188 Diabète athérothrombose Thérapies Réunion Océan Indien (DéTROI), F-97490 Sainte-Clotilde, France; [email protected] (S.T.); [email protected] (D.C.) 2 CHU de La Réunion, Centre d’Investigations Clinique 1410, 97410 Saint-Pierre, France 3 AP-HP, Service d’Anesthésie-Réanimation, CHU Bichat-Claude Bernard, 75018 Paris, France 4 CHU de La Réunion, Neurocritical Care Unit, 97410 Saint-Pierre, France * Correspondence: [email protected]; Tel.: +33-262-93-88-11 Received: 7 March 2020; Accepted: 6 April 2020; Published: 12 April 2020 Abstract: Lipoproteins were initially defined according to their composition (lipids and proteins) and classified according to their density (from very low- to high-density lipoproteins—HDLs). Whereas their capacity to transport hydrophobic lipids in a hydrophilic environment (plasma) is not questionable, their primitive function of cholesterol transporter could be challenged. All lipoproteins are reported to bind and potentially neutralize bacterial lipopolysaccharides (LPS); this is particularly true for HDL particles. In addition, HDL levels are drastically decreased under infectious conditions such as sepsis, suggesting a potential role in the clearance of bacterial material and, particularly, LPS. Moreover, “omics” technologies have unveiled significant changes in HDL composition in different inflammatory states, ranging from acute inflammation occurring during septic shock to low-grade inflammation associated with moderate endotoxemia such as periodontal disease or obesity. In this review, we will discuss HDL modifications associated with exposure to pathogens including bacteria, viruses and parasites, with a special focus on sepsis and the potential of HDL therapy in this context. -

Association Between the APOA2 Rs3813627 Single Nucleotide Polymorphism and HDL and APOA1 Levels Through BMI
biomedicines Article Association between the APOA2 rs3813627 Single Nucleotide Polymorphism and HDL and APOA1 Levels Through BMI Hatim Boughanem 1 , Borja Bandera-Merchán 2, Pablo Hernández-Alonso 2,3,4 , Noelia Moreno-Morales 5, Francisco José Tinahones 2,3, José Lozano 6 , Sonsoles Morcillo 2,3,* and Manuel Macias-Gonzalez 2,3,* 1 Instituto de Investigación Biomédica de Málaga (IBIMA), Facultad de Ciencias, Universidad de Málaga, 29010 Málaga, Spain; [email protected] 2 Unidad de Gestión Clínica de Endocrinología y Nutrición del Hospital Virgen de la Victoria, Instituto de Investigación Biomédica de Málaga (IBIMA), Universidad de Málaga, 29010 Málaga, Spain; [email protected] (B.B.-M.); [email protected] (P.H.-A.); [email protected] (F.J.T.) 3 Centro de Investigación Biomédica en Red de Fisiopatología de la Obesidad y la Nutrición, CIBERObn, 28029 Madrid, Spain 4 Human Nutrition Unit, Faculty of Medicine and Health Sciences, Sant Joan Hospital, Institut d’Investigació Sanitària Pere Virgili, Rovira i Virgili University, 43201 Reus, Spain 5 Department of Physiotherapy, School of Health Sciences, University of Malaga-Instituto de Investigación Biomédica de Málaga (IBIMA), 29010 Málaga, Spain; [email protected] 6 Departamento de Bioquímica y Biología Molecular, Facultad de Ciencias, Universidad de Málaga, 29010 Málaga, Spain; [email protected] * Correspondence: [email protected] (S.M.); [email protected] (M.M.-G.); Tel.: +34-951-032-648 (S.M. & M.M.-G.); Fax: +34-27-951-924-651 (S.M. & M.M.-G.) Received: 18 February 2020; Accepted: 25 February 2020; Published: 27 February 2020 Abstract: Background: The interaction between obesity and genetic traits on high density lipoprotein (HDL) levels has been extensively studied. -

Downloaded Separately for CEU (Individuals of Western and Northern European Origin) and YRI (Yoruba in Nigeria) from Hapmap Phase 2
NIH Public Access Author Manuscript Int J Obes (Lond). Author manuscript; available in PMC 2013 September 11. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Int J Obes (Lond). 2013 September ; 37(9): 1211–1220. doi:10.1038/ijo.2012.215. Lipoprotein receptor-related protein 1 variants and dietary fatty acids: meta-analysis of European origin and African American studies CE Smith1, J Ngwa2, T Tanaka3, Q Qi4, MK Wojczynski5, RN Lemaitre6, JS Anderson7, A Manichaikul8, V Mikkilä9, FJA van Rooij10,11, Z Ye12, S Bandinelli13, AC Frazier-Wood14, DK Houston15, F Hu4,16, C Langenberg12, NM McKeown1, D Mozaffarian17,18, KE North19, J Viikari20, MC Zillikens11,21, L Djoussé22, A Hofman10,11, M Kähönen23, EK Kabagambe14, RJF Loos12, GB Saylor7, NG Forouhi12, Y Liu24, KJ Mukamal25, Y-DI Chen26, MY Tsai27, AG Uitterlinden10,11,21, O Raitakari28, CM van Duijn10,11, DK Arnett14, IB Borecki5, LA Cupples2,29, L Ferrucci3, SB Kritchevsky15, T Lehtimäki30, Lu Qi4,16, JI Rotter26, DS Siscovick31, NJ Wareham12, JCM Witteman10,11, JM Ordovás1,32,33, and JA Nettleton34 1Nutrition and Genomics Laboratory, Jean Mayer USDA Human Nutrition Research Center on Aging, Tufts University, Boston, MA, USA 2Department of Biostatistics, Boston University School of Public Health, Boston, MA, USA 3Clinical Research Branch, National Institute on Aging, Baltimore, MD, USA 4Department of Nutrition, Harvard School of Public Health, Boston, MA, USA 5Department of Genetics, Washington University School of Medicine, St. -

MAP17's Up-Regulation, a Crosspoint in Cancer and Inflammatory
García-Heredia and Carnero Molecular Cancer (2018) 17:80 https://doi.org/10.1186/s12943-018-0828-7 REVIEW Open Access Dr. Jekyll and Mr. Hyde: MAP17’s up- regulation, a crosspoint in cancer and inflammatory diseases José M. García-Heredia1,2,3 and Amancio Carnero1,3* Abstract Inflammation is a common defensive response that is activated after different harmful stimuli. This chronic, or pathological, inflammation is also one of the causes of neoplastic transformation and cancer development. MAP17 is a small protein localized to membranes with a restricted pattern of expression in adult tissues. However, its expression is common in destabilized cells, as it is overexpressed both in inflammatory diseases and in cancer. MAP17 is overexpressed in most, if not all, carcinomas and in many tumors of mesenchymal origin, and correlates with higher grade and poorly differentiated tumors. This overexpression drives deep changes in cell homeostasis including increased oxidative stress, deregulation of signaling pathways and increased growth rates. Importantly, MAP17 is associated in tumors with inflammatory cells infiltration, not only in cancer but in various inflammatory diseases such as Barret’s esophagus, lupus, Crohn’s, psoriasis and COPD. Furthermore, MAP17 also modifies the expression of genes connected to inflammation, showing a clear induction of the inflammatory profile. Since MAP17 appears highly correlated with the infiltration of inflammatory cells in cancer, is MAP17 overexpression an important cellular event connecting tumorigenesis and inflammation? Keywords: MAP17, Cancer, Inflammatory diseases Background process ends when the activated cells undergo apoptosis in Inflammatory response is a common defensive process acti- a highly regulated process that finishes after pathogens and vated after different harmful stimuli, constituting a highly cell debris have been phagocytized [9]. -

Common Genetic Variations Involved in the Inter-Individual Variability Of
nutrients Review Common Genetic Variations Involved in the Inter-Individual Variability of Circulating Cholesterol Concentrations in Response to Diets: A Narrative Review of Recent Evidence Mohammad M. H. Abdullah 1 , Itzel Vazquez-Vidal 2, David J. Baer 3, James D. House 4 , Peter J. H. Jones 5 and Charles Desmarchelier 6,* 1 Department of Food Science and Nutrition, Kuwait University, Kuwait City 10002, Kuwait; [email protected] 2 Richardson Centre for Functional Foods & Nutraceuticals, University of Manitoba, Winnipeg, MB R3T 6C5, Canada; [email protected] 3 United States Department of Agriculture, Agricultural Research Service, Beltsville, MD 20705, USA; [email protected] 4 Department of Food and Human Nutritional Sciences, University of Manitoba, Winnipeg, MB R3T 2N2, Canada; [email protected] 5 Nutritional Fundamentals for Health, Vaudreuil-Dorion, QC J7V 5V5, Canada; [email protected] 6 Aix Marseille University, INRAE, INSERM, C2VN, 13005 Marseille, France * Correspondence: [email protected] Abstract: The number of nutrigenetic studies dedicated to the identification of single nucleotide Citation: Abdullah, M.M.H.; polymorphisms (SNPs) modulating blood lipid profiles in response to dietary interventions has Vazquez-Vidal, I.; Baer, D.J.; House, increased considerably over the last decade. However, the robustness of the evidence-based sci- J.D.; Jones, P.J.H.; Desmarchelier, C. ence supporting the area remains to be evaluated. The objective of this review was to present Common Genetic Variations Involved recent findings concerning the effects of interactions between SNPs in genes involved in cholesterol in the Inter-Individual Variability of metabolism and transport, and dietary intakes or interventions on circulating cholesterol concen- Circulating Cholesterol trations, which are causally involved in cardiovascular diseases and established biomarkers of Concentrations in Response to Diets: cardiovascular health. -
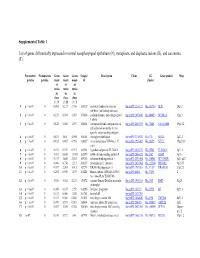
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

Prenatal Detection of TAR Syndrome in a Fetus with Compound Inheritance of an RBM8A SNP and a 334‑Kb Deletion: a Case Report
MOLECULAR MEDICINE REPORTS 9: 163-165, 2014 Prenatal detection of TAR syndrome in a fetus with compound inheritance of an RBM8A SNP and a 334‑kb deletion: A case report IOANNIS PAPOULIDIS1, EIRINI OIKONOMIDOU1, SANDRO ORRU2, ELISAVET SIOMOU1, MARIA KONTODIOU1, MAKARIOS ELEFTHERIADES3, VASILIOS BACOULAS4, JUAN C. CIGUDOSA5, JAVIER SUELA5, LORETTA THOMAIDIS6 and EMMANOUIL MANOLAKOS1,2 1Laboratory of Genetics, Eurogenetica S.A., Thessaloniki 55133, Greece; 2Department of Medical Genetics, University of Cagliari, Binaghi Hospital, Cagliari I‑09126, Italy; 3Embryocare, Fetal Medicine Unit, Athens 11522; 4Fetal Medicine Centre, Athens 10674, Greece; 5NIMGenetics, Madrid 28049, Spain; 6Department of Pediatrics, Aglaia Kyriakou Children's Hospital, University of Athens, Athens 11527, Greece Received May 30, 2013; Accepted October 16, 2013 DOI: 10.3892/mmr.2013.1788 Abstract. Thrombocytopenia-absent radius syndrome identified the presence of a minimally deleted 200‑kb region (TAR) is a rare genetic disorder that is characterized by the at chromosome band 1q21.1 in patients with TAR, but it is not absence of the radius bone in each forearm and a markedly sufficient to cause the phenotype (3,4). A study identified two reduced platelet count that results in life-threatening bleeding rare single nucleotide polymorphisms (SNPs) in the regulatory episodes (thrombocytopenia). Tar syndrome has been associ- region of the RBM8A gene that are involved in TAR syndrome ated with a deletion of a segment of 1q21.1 cytoband. The through the reduction of the expression of the RBM8A-encoded 1q21.1 deletion syndrome phenotype includes Tar and other Y14 protein (4). The first allele (rs139428292 G>A), which is features such as mental retardation, autism and microcephaly. -

Supplementary Table S1. List of Differentially Expressed
Supplementary table S1. List of differentially expressed transcripts (FDR adjusted p‐value < 0.05 and −1.4 ≤ FC ≥1.4). 1 ID Symbol Entrez Gene Name Adj. p‐Value Log2 FC 214895_s_at ADAM10 ADAM metallopeptidase domain 10 3,11E‐05 −1,400 205997_at ADAM28 ADAM metallopeptidase domain 28 6,57E‐05 −1,400 220606_s_at ADPRM ADP‐ribose/CDP‐alcohol diphosphatase, manganese dependent 6,50E‐06 −1,430 217410_at AGRN agrin 2,34E‐10 1,420 212980_at AHSA2P activator of HSP90 ATPase homolog 2, pseudogene 6,44E‐06 −1,920 219672_at AHSP alpha hemoglobin stabilizing protein 7,27E‐05 2,330 aminoacyl tRNA synthetase complex interacting multifunctional 202541_at AIMP1 4,91E‐06 −1,830 protein 1 210269_s_at AKAP17A A‐kinase anchoring protein 17A 2,64E‐10 −1,560 211560_s_at ALAS2 5ʹ‐aminolevulinate synthase 2 4,28E‐06 3,560 212224_at ALDH1A1 aldehyde dehydrogenase 1 family member A1 8,93E‐04 −1,400 205583_s_at ALG13 ALG13 UDP‐N‐acetylglucosaminyltransferase subunit 9,50E‐07 −1,430 207206_s_at ALOX12 arachidonate 12‐lipoxygenase, 12S type 4,76E‐05 1,630 AMY1C (includes 208498_s_at amylase alpha 1C 3,83E‐05 −1,700 others) 201043_s_at ANP32A acidic nuclear phosphoprotein 32 family member A 5,61E‐09 −1,760 202888_s_at ANPEP alanyl aminopeptidase, membrane 7,40E‐04 −1,600 221013_s_at APOL2 apolipoprotein L2 6,57E‐11 1,600 219094_at ARMC8 armadillo repeat containing 8 3,47E‐08 −1,710 207798_s_at ATXN2L ataxin 2 like 2,16E‐07 −1,410 215990_s_at BCL6 BCL6 transcription repressor 1,74E‐07 −1,700 200776_s_at BZW1 basic leucine zipper and W2 domains 1 1,09E‐06 −1,570 222309_at -

Role of Myeloperoxidase Mediated Oxidative Modification and Apolipoprotein Composition in High Density Lipoprotein Function
ROLE OF MYELOPEROXIDASE MEDIATED OXIDATIVE MODIFICATION AND APOLIPOPROTEIN COMPOSITION IN HIGH DENSITY LIPOPROTEIN FUNCTION by ARUNDHATI UNDURTI Submitted in partial fulfillment of the requirements For the degree of Doctor of Philosophy Thesis Advisor: Dr. Stanley L. Hazen Department of Microbiology and Molecular Biology Cell Biology Program CASE WESTERN RESERVE UNIVERSITY August, 2010 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of _____________________________________________________Arundhati Undurti candidate for the ______________________degreePhD *. Alan Levine (signed)_______________________________________________ (chair of the committee) Stanley Hazen ________________________________________________ Jonathan Smith ________________________________________________ Menachem Shoham ________________________________________________ Mark Chance ________________________________________________ ________________________________________________ (date) _______________________06-30-2010 *We also certify that written approval has been obtained for any proprietary material contained therein. For Amma and Nana TABLE OF CONTENTS List of Figures 3 List of Tables 7 Abbreviations 8 Acknowledgements 11 Abstract 13 Chapter I: Introduction Pathogenesis of Atherosclerosis Endothelial Dysfunction 17 Fatty Streak Formation 17 Advanced Lesion Formation 18 Thrombotic Complications 18 Role of Lipoproteins in Atherosclerosis Lipoprotein Classification and Metabolism 19 High Density Lipoprotein 20 Reverse