Iron Supplementation Regulates the Progression of High Fat Diet Induced
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

The Title of the Dissertation
UNIVERSITY OF CALIFORNIA SAN DIEGO Novel network-based integrated analyses of multi-omics data reveal new insights into CD8+ T cell differentiation and mouse embryogenesis A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioinformatics and Systems Biology by Kai Zhang Committee in charge: Professor Wei Wang, Chair Professor Pavel Arkadjevich Pevzner, Co-Chair Professor Vineet Bafna Professor Cornelis Murre Professor Bing Ren 2018 Copyright Kai Zhang, 2018 All rights reserved. The dissertation of Kai Zhang is approved, and it is accept- able in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California San Diego 2018 iii EPIGRAPH The only true wisdom is in knowing you know nothing. —Socrates iv TABLE OF CONTENTS Signature Page ....................................... iii Epigraph ........................................... iv Table of Contents ...................................... v List of Figures ........................................ viii List of Tables ........................................ ix Acknowledgements ..................................... x Vita ............................................. xi Abstract of the Dissertation ................................. xii Chapter 1 General introduction ............................ 1 1.1 The applications of graph theory in bioinformatics ......... 1 1.2 Leveraging graphs to conduct integrated analyses .......... 4 1.3 References .............................. 6 Chapter 2 Systematic -

Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle
Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle Slim Ben-Jemaa, Gabriele Senczuk, Elena Ciani, Roberta Ciampolini, Gennaro Catillo, Mekki Boussaha, Fabio Pilla, Baldassare Portolano, Salvatore Mastrangelo To cite this version: Slim Ben-Jemaa, Gabriele Senczuk, Elena Ciani, Roberta Ciampolini, Gennaro Catillo, et al.. Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle. Frontiers in Genetics, Frontiers, 2021, 10.3389/fgene.2021.675569. hal-03210766 HAL Id: hal-03210766 https://hal.inrae.fr/hal-03210766 Submitted on 28 Apr 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License ORIGINAL RESEARCH published: 28 April 2021 doi: 10.3389/fgene.2021.675569 Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle Slim Ben-Jemaa 1, Gabriele Senczuk 2, Elena Ciani 3, Roberta -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Nucleic Acid High-Throughput Sequencing Studies Present Unique Challenges in Analysis and Interpretation
Nucleic Acid High-Throughput Sequencing Studies Present Unique Challenges in Analysis and Interpretation DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Kenji Oman, B.S., M.S. Graduate Program in Physics The Ohio State University 2015 Dissertation Committee: Dr. Ralf Bundschuh, Advisor Dr. Kurt Fredrick Dr. Richard Furnstahl Dr. Michael Poirier c Copyright by Kenji Oman 2015 Abstract From the discovery of nucleic acids, their significance as an information carrier in the cell, and with the development of high-throughput sequencing (HTS) techniques, molecular biology has seen ever-increasing developments in our understanding of the mechanisms of life. Here we first present a small overview of the progression of our understanding of nucleic acids, and current HTS techniques used to study them. We then investigate the interaction of methyl-binding-domain (MBD) with methylated DNA, as used in MethylCap-seq (a HTS technique), and present a model for their interaction, and a Bayesian model utilizing our increased understanding to predict methylation levels in samples with an unknown methylation profile. We next introduce a HTS analysis pipeline we have developed, and examine the use of this pipeline in the analysis of 5′-end seq data, ultimately leading to its abandonment. Finally, we present a further application of the pipeline in our investigation of lepA’s role in translation initiation and elongation in E. coli. ii To my parents, who always told me I could. iii Acknowledgments There are many people who have helped me get to the point of a Ph.D. -

Regulation of Adult Neurogenesis in Mammalian Brain
International Journal of Molecular Sciences Review Regulation of Adult Neurogenesis in Mammalian Brain 1,2, 3, 3,4 Maria Victoria Niklison-Chirou y, Massimiliano Agostini y, Ivano Amelio and Gerry Melino 3,* 1 Centre for Therapeutic Innovation (CTI-Bath), Department of Pharmacy & Pharmacology, University of Bath, Bath BA2 7AY, UK; [email protected] 2 Blizard Institute of Cell and Molecular Science, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London E1 2AT, UK 3 Department of Experimental Medicine, TOR, University of Rome “Tor Vergata”, 00133 Rome, Italy; [email protected] (M.A.); [email protected] (I.A.) 4 School of Life Sciences, University of Nottingham, Nottingham NG7 2HU, UK * Correspondence: [email protected] These authors contributed equally to this work. y Received: 18 May 2020; Accepted: 7 July 2020; Published: 9 July 2020 Abstract: Adult neurogenesis is a multistage process by which neurons are generated and integrated into existing neuronal circuits. In the adult brain, neurogenesis is mainly localized in two specialized niches, the subgranular zone (SGZ) of the dentate gyrus and the subventricular zone (SVZ) adjacent to the lateral ventricles. Neurogenesis plays a fundamental role in postnatal brain, where it is required for neuronal plasticity. Moreover, perturbation of adult neurogenesis contributes to several human diseases, including cognitive impairment and neurodegenerative diseases. The interplay between extrinsic and intrinsic factors is fundamental in regulating neurogenesis. Over the past decades, several studies on intrinsic pathways, including transcription factors, have highlighted their fundamental role in regulating every stage of neurogenesis. However, it is likely that transcriptional regulation is part of a more sophisticated regulatory network, which includes epigenetic modifications, non-coding RNAs and metabolic pathways. -

Predicting Gene Ontology Biological Process from Temporal Gene Expression Patterns Astrid Lægreid,1,4 Torgeir R
Methods Predicting Gene Ontology Biological Process From Temporal Gene Expression Patterns Astrid Lægreid,1,4 Torgeir R. Hvidsten,2 Herman Midelfart,2 Jan Komorowski,2,3,4 and Arne K. Sandvik1 1Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology, N-7489 Trondheim, Norway; 2Department of Information and Computer Science, Norwegian University of Science and Technology, N-7491 Trondheim, Norway; 3The Linnaeus Centre for Bioinformatics, Uppsala University, SE-751 24 Uppsala, Sweden The aim of the present study was to generate hypotheses on the involvement of uncharacterized genes in biological processes. To this end,supervised learning was used to analyz e microarray-derived time-series gene expression data. Our method was objectively evaluated on known genes using cross-validation and provided high-precision Gene Ontology biological process classifications for 211 of the 213 uncharacterized genes in the data set used. In addition,new roles in biological process were hypothesi zed for known genes. Our method uses biological knowledge expressed by Gene Ontology and generates a rule model associating this knowledge with minimal characteristic features of temporal gene expression profiles. This model allows learning and classification of multiple biological process roles for each gene and can predict participation of genes in a biological process even though the genes of this class exhibit a wide variety of gene expression profiles including inverse coregulation. A considerable number of the hypothesized new roles for known genes were confirmed by literature search. In addition,many biological process roles hypothesi zed for uncharacterized genes were found to agree with assumptions based on homology information. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

The Suppressive Effects of 1,25-Dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 8-2014 The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration Carolyn Jeanne Ricciardi University of Tennessee - Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Part of the Molecular, Genetic, and Biochemical Nutrition Commons Recommended Citation Ricciardi, Carolyn Jeanne, "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration. " Master's Thesis, University of Tennessee, 2014. https://trace.tennessee.edu/utk_gradthes/2876 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Carolyn Jeanne Ricciardi entitled "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Master of Science, with a major in Nutrition. -

COX6B1 Antibody - N-Terminal Region Rabbit Polyclonal Antibody Catalog # AI15391
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 COX6B1 Antibody - N-terminal region Rabbit Polyclonal Antibody Catalog # AI15391 Specification COX6B1 Antibody - N-terminal region - Product Information Application WB Primary Accession P14854 Other Accession NM_001863, NP_001854 Reactivity Human, Mouse, Rat, Rabbit, Pig, Horse, Yeast, Bovine, Guinea Pig, Dog Predicted Human, Mouse, Host: Rabbit Rat, Rabbit, Pig, Target Name: COX6B1 Horse, Yeast, Sample Tissue: OVCAR-3 Whole cell lysate Bovine, Guinea Pig, Dog s Host Rabbit Antibody Dilution: 1.0μg/ml Clonality Polyclonal Calculated MW 10kDa KDa COX6B1 Antibody - N-terminal region - References COX6B1 Antibody - N-terminal region - Additional Information Taanman J.-W.,et al.Nucleic Acids Res. 17:1766-1766(1989). Gene ID 1340 Taanman J.-W.,et al.Gene 93:285-291(1990). Carrero-Valenzuela R.D.,et al.Gene Alias Symbol COX6B, COXG, 102:229-236(1991). COXVIb1 Ota T.,et al.Nat. Genet. 36:40-45(2004). Other Names Kalnine N.,et al.Submitted (MAY-2003) to the Cytochrome c oxidase subunit 6B1, EMBL/GenBank/DDBJ databases. Cytochrome c oxidase subunit VIb isoform 1, COX VIb-1, COX6B1, COX6B Format Liquid. Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and 2% sucrose. Reconstitution & Storage Add 50 ul of distilled water. Final anti-COX6B1 antibody concentration is 1 mg/ml in PBS buffer with 2% sucrose. For longer periods of storage, store at 20°C. Avoid repeat freeze-thaw cycles. Precautions COX6B1 Antibody - N-terminal region is for Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 research use only and not for use in diagnostic or therapeutic procedures. -
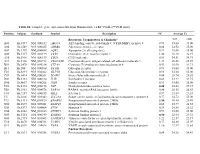
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90 -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo