UCLA Electronic Theses and Dissertations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Abstracts Genome 10K & Genome Science 29 Aug - 1 Sept 2017 Norwich Research Park, Norwich, Uk
Genome 10K c ABSTRACTS GENOME 10K & GENOME SCIENCE 29 AUG - 1 SEPT 2017 NORWICH RESEARCH PARK, NORWICH, UK Genome 10K c 48 KEYNOTE SPEAKERS ............................................................................................................................... 1 Dr Adam Phillippy: Towards the gapless assembly of complete vertebrate genomes .................... 1 Prof Kathy Belov: Saving the Tasmanian devil from extinction ......................................................... 1 Prof Peter Holland: Homeobox genes and animal evolution: from duplication to divergence ........ 2 Dr Hilary Burton: Genomics in healthcare: the challenges of complexity .......................................... 2 INVITED SPEAKERS ................................................................................................................................. 3 Vertebrate Genomics ........................................................................................................................... 3 Alex Cagan: Comparative genomics of animal domestication .......................................................... 3 Plant Genomics .................................................................................................................................... 4 Ksenia Krasileva: Evolution of plant Immune receptors ..................................................................... 4 Andrea Harper: Using Associative Transcriptomics to predict tolerance to ash dieback disease in European ash trees ............................................................................................................ -

3718 Issue63july2010 1.Pdf
Issue 63.qxd:Genetic Society News 1/10/10 14:41 Page 1 JULYJULLYY 2010 | ISSUEISSUE 63 GENETICSGENNETICSS SOCIETYSOCIEETY NENEWSEWS In this issue The Genetics Society NewsNewws is edited by U Genetics Society PresidentPresident Honoured Honoured ProfProf David Hosken and items ittems for future future issues can be sent to thee editor,editor, preferably preferably U Mouse Genetics Meeting by email to [email protected],D.J.Hosken@@exeter.ac.uk, or U SponsoredSponsored Meetings Meetings hardhard copy to Chair in Evolutionary Evoolutionary Biology, Biology, UniversityUniversity of Exeter,Exeter, Cornwall Cornnwall Campus, U The JBS Haldane LectureLecture Tremough,Tremough, Penryn, TR10 0 9EZ UK.UK. The U Schools Evolutionn ConferenceConference Newsletter is published twicet a year,year, with copy dates of 1st June andand 26th November.November. U TaxiTaxi Drivers The British YeastYeaste Group Group descend on Oxford Oxford for their 2010 meeting: m see the reportreport on page 35. 3 Image © Georgina McLoughlin Issue 63.qxd:Genetic Society News 1/10/10 14:41 Page 2 A WORD FROM THE EDITOR A word from the editor Welcome to issue 63. In this issue we announce a UK is recognised with the award of a CBE in the new Genetics Society Prize to Queen’s Birthday Honours, tells us about one of Welcome to my last issue as join the medals and lectures we her favourite papers by Susan Lindquist, the 2010 editor of the Genetics Society award. The JBS Haldane Mendel Lecturer. Somewhat unusually we have a News, after 3 years in the hot Lecture will be awarded couple of Taxi Drivers in this issue – Brian and seat and a total of 8 years on annually to recognise Deborah Charlesworth are not so happy about the committee it is time to excellence in communicating the way that the print media deals with some move on before I really outstay aspects of genetics research to scientific issues and Chris Ponting bemoans the my welcome! It has been a the public. -

Exploiting High Throughput DNA Sequencing Data for Genomic Analysis
Exploiting high throughput DNA sequencing data for genomic analysis Markus Hsi-Yang Fritz Darwin College A dissertation submitted to the University of Cambridge for the degree of Doctor of Philosophy 14th October 2011 Markus Hsi-Yang Fritz EMBL-European Bioinformatics Institute Wellcome Trust Genome Campus Hinxton, Cambridge, CB10 1SD United Kingdom email: [email protected] This dissertation is the result of my own work and includes nothing which is the outcome of work done in collaboration except where specifically indicated in the text. No part of this work has been submitted or is currently being submitted for any other qualification. This document does not exceed the word limit of 60,000 words1 as defined by the Biology Degree Committee. Markus Hsi-Yang Fritz 14th October 2011 1 excluding bibliography, figures, appendices etc. To an exceptional scientist — my Dad. Exploiting high throughput DNA sequencing data for genomic analysis Summary Markus Hsi-Yang Fritz Darwin College The last few years have witnessed a drastic increase in genomic data. This has been facilitated by the shift away from the Sanger sequencing tech- nique to an array of high-throughput methods — so-called next-generation sequencing technologies. This enormous growth of available DNA data has been a tremendous boon to large-scale genomics studies and has rapidly advanced fields such as environmental genomics, ancient DNA research, population genomics and disease association. On the other hand, however, researchers and sequence archives are now facing an enormous data deluge. Critically, the rate of sequencing data accumulation is now outstripping advances in hard drive capacity, network bandwidth and processing power. -

Strategic Plan 2011-2016
Strategic Plan 2011-2016 Wellcome Trust Sanger Institute Strategic Plan 2011-2016 Mission The Wellcome Trust Sanger Institute uses genome sequences to advance understanding of the biology of humans and pathogens in order to improve human health. -i- Wellcome Trust Sanger Institute Strategic Plan 2011-2016 - ii - Wellcome Trust Sanger Institute Strategic Plan 2011-2016 CONTENTS Foreword ....................................................................................................................................1 Overview .....................................................................................................................................2 1. History and philosophy ............................................................................................................ 5 2. Organisation of the science ..................................................................................................... 5 3. Developments in the scientific portfolio ................................................................................... 7 4. Summary of the Scientific Programmes 2011 – 2016 .............................................................. 8 4.1 Cancer Genetics and Genomics ................................................................................ 8 4.2 Human Genetics ...................................................................................................... 10 4.3 Pathogen Variation .................................................................................................. 13 4.4 Malaria -

Pnas11052ackreviewers 5098..5136
Acknowledgment of Reviewers, 2013 The PNAS editors would like to thank all the individuals who dedicated their considerable time and expertise to the journal by serving as reviewers in 2013. Their generous contribution is deeply appreciated. A Harald Ade Takaaki Akaike Heather Allen Ariel Amir Scott Aaronson Karen Adelman Katerina Akassoglou Icarus Allen Ido Amit Stuart Aaronson Zach Adelman Arne Akbar John Allen Angelika Amon Adam Abate Pia Adelroth Erol Akcay Karen Allen Hubert Amrein Abul Abbas David Adelson Mark Akeson Lisa Allen Serge Amselem Tarek Abbas Alan Aderem Anna Akhmanova Nicola Allen Derk Amsen Jonathan Abbatt Neil Adger Shizuo Akira Paul Allen Esther Amstad Shahal Abbo Noam Adir Ramesh Akkina Philip Allen I. Jonathan Amster Patrick Abbot Jess Adkins Klaus Aktories Toby Allen Ronald Amundson Albert Abbott Elizabeth Adkins-Regan Muhammad Alam James Allison Katrin Amunts Geoff Abbott Roee Admon Eric Alani Mead Allison Myron Amusia Larry Abbott Walter Adriani Pietro Alano Isabel Allona Gynheung An Nicholas Abbott Ruedi Aebersold Cedric Alaux Robin Allshire Zhiqiang An Rasha Abdel Rahman Ueli Aebi Maher Alayyoubi Abigail Allwood Ranjit Anand Zalfa Abdel-Malek Martin Aeschlimann Richard Alba Julian Allwood Beau Ances Minori Abe Ruslan Afasizhev Salim Al-Babili Eric Alm David Andelman Kathryn Abel Markus Affolter Salvatore Albani Benjamin Alman John Anderies Asa Abeliovich Dritan Agalliu Silas Alben Steven Almo Gregor Anderluh John Aber David Agard Mark Alber Douglas Almond Bogi Andersen Geoff Abers Aneel Aggarwal Reka Albert Genevieve Almouzni George Andersen Rohan Abeyaratne Anurag Agrawal R. Craig Albertson Noga Alon Gregers Andersen Susan Abmayr Arun Agrawal Roy Alcalay Uri Alon Ken Andersen Ehab Abouheif Paul Agris Antonio Alcami Claudio Alonso Olaf Andersen Soman Abraham H. -

Current Positions
Jay Shendure, MD, PhD Updated December 31, 2020 Current Positions Investigator, Howard Hughes Medical Institute Professor, Genome Sciences, University of Washington Director, Allen Discovery Center for Cell Lineage Director, Brotman Baty Institute for Precision Medicine Contact Information E-mail: [email protected] Lab website: http://krishna.gs.washington.edu Office phone: (206) 685-8543 Education • 2007 M.D., Harvard Medical School (Boston, Massachusetts) • 2005 Ph.D. in Genetics, Harvard University (Cambridge, Massachusetts) Research Advisor: George M. Church Thesis entitled “Multiplex Genome Sequencing and Analysis” • 1996 A.B., summa cum laude in Molecular Biology, Princeton University (Princeton, NJ) Research Advisor: Lee M. Silver Professional Experience • 2017 – present Scientific Director Brotman-Baty Institute for Precision Medicine • 2017 – present Scientific Director Allen Discovery Center for Cell Lineage Tracing • 2015 – present Investigator Howard Hughes Medical Institute • 2015 – present Full Professor (with tenure) Department of Genome Sciences, University of Washington, Seattle, WA • 2010 – present Affiliate Professor Division of Human Biology, Fred Hutchinson Cancer Research Center, Seattle, WA • 2011 – 2015 Associate Professor (with tenure) Department of Genome Sciences, University of Washington, Seattle, WA • 2007 – 2011 Assistant Professor Department of Genome Sciences, University of Washington, Seattle, WA • 1998 – 2007 Medical Scientist Training Program (MSTP) Candidate Department of Genetics, Harvard Medical School, Boston, WA • 1997 – 1998 Research Scientist Vaccine Division, Merck Research Laboratories, Rahway, NJ • 1996 – 1997 Fulbright Scholar to India Department of Pediatrics, Sassoon General Hospital, Pune, India 1 Jay Shendure, MD, PhD Honors, Awards, Named Lectures • 2019 Richard Lounsbery Award (for extraordinary scientific achievement in biology & medicine) National Academy of Sciences • 2019 Jeffrey M. Trent Lectureship in Cancer Research National Human Genome Research Institute, National Institutes of Health • 2019 Paul D. -

New Experimental Evidence and Its Consequences for Linguistics
Genetic influences on tone? New experimental evidence and its consequences for linguistics. Dan Dediu1* and D. Robert Ladd2 1 Laboratoire Dynamique Du Langage UMR 5596, Université Lumière Lyon 2, Lyon, France 2 Linguistics and English Language, The University of Edinburgh, Edinburgh, UK * Corresponding author: [email protected] Abstract: In 2007, we used a cross-linguistic statistical analysis to show that there seems to be a negative correlation between the population frequency of the “derived” alleles of two genes involved in brain growth and development, ASPM and Microcephalin (or MCHP1), and the use of tone in the language(s) spoken by the population. Despite our best attempts at disentangling the confounding effects of contact and genealogical inheritance, and the fact that these correlations stood out among millions of similar correlations between multiple genetic markers and linguistic features, this was, at best, a statistical association and, at worst, a false positive. However, new experimental evidence has recently been published independently of us, showing that the negative correlation between the “derived” allele of ASPM and tone perception and/or processing is present at the level of inter-individual differences among more than 400 native speakers of Cantonese, providing strong support for our initial findings (on the other hand, there is no signal for MCPH1). Here we contextualize and summarize these experimental findings, we address some remaining puzzles (including MCHP1 and the absence of tone in Australia), and we discuss the implications of these results for the language sciences. Genes & tone: a comment on new experimental evidence and its consequences Introduction: genetic influences on language and speech At the most general level, it is beyond debate that there is something about the human genome that makes language possible. -

BIOLOGY 639 SCIENCE ONLINE the Unexpected Brains Behind Blood Vessel Growth 641 THIS WEEK in SCIENCE 668 U.K
4 February 2005 Vol. 307 No. 5710 Pages 629–796 $10 07%.'+%#%+& 2416'+0(70%6+10 37#06+6#6+8' 51(69#4' #/2.+(+%#6+10 %'..$+1.1); %.10+0) /+%41#44#;5 #0#.;5+5 #0#.;5+5 2%4 51.76+105 Finish first with a superior species. 50% faster real-time results with FullVelocity™ QPCR Kits! Our FullVelocity™ master mixes use a novel enzyme species to deliver Superior Performance vs. Taq -Based Reagents FullVelocity™ Taq -Based real-time results faster than conventional reagents. With a simple change Reagent Kits Reagent Kits Enzyme species High-speed Thermus to the thermal profile on your existing real-time PCR system, the archaeal Fast time to results FullVelocity technology provides you high-speed amplification without Enzyme thermostability dUTP incorporation requiring any special equipment or re-optimization. SYBR® Green tolerance Price per reaction $$$ • Fast, economical • Efficient, specific and • Probe and SYBR® results sensitive Green chemistries Need More Information? Give Us A Call: Ask Us About These Great Products: Stratagene USA and Canada Stratagene Europe FullVelocity™ QPCR Master Mix* 600561 Order: (800) 424-5444 x3 Order: 00800-7000-7000 FullVelocity™ QRT-PCR Master Mix* 600562 Technical Services: (800) 894-1304 Technical Services: 00800-7400-7400 FullVelocity™ SYBR® Green QPCR Master Mix 600581 FullVelocity™ SYBR® Green QRT-PCR Master Mix 600582 Stratagene Japan K.K. *U.S. Patent Nos. 6,528,254, 6,548,250, and patents pending. Order: 03-5159-2060 Purchase of these products is accompanied by a license to use them in the Polymerase Chain Reaction (PCR) Technical Services: 03-5159-2070 process in conjunction with a thermal cycler whose use in the automated performance of the PCR process is YYYUVTCVCIGPGEQO covered by the up-front license fee, either by payment to Applied Biosystems or as purchased, i.e., an authorized thermal cycler. -

Job Description and Person Specificationselection Criteria
_________________________________________________________________________ Job Description Florence Nightingale Bicentennial Fellowship and Tutor in Statistics Job title and Probability Division Mathematical, Physical and Life Sciences Department Statistics Location 24-29 St Giles’, Oxford, OX1 3LB Grade and salary Grade 8: £40,792 - £48,677 per annum Hours Full time Contract type Fixed-term until 30 September 2024 Reporting to Head of Department Vacancy reference 141486 The role The Department of Statistics is recruiting a Florence Nightingale Bicentennial Fellowship and Tutor in Statistics and Probability with effect from October 2019 or as soon as possible thereafter. The post holder will join the dynamic and collaborative Department of Statistics. The Department carries out world-leading research in applied statistics fields including statistical and population genetics and bioinformatics, as well as core theoretical statistics, computational statistics, machine learning and probability. This is an exciting time for the Department, which relocated to new premises on St Giles’ in the heart of the University of Oxford in 2015. Our newly-renovated building provides state-of-the-art teaching facilities and modern space to facilitate collaboration and integration, creating a highly visible centre for Statistics in Oxford. The successful candidate will hold a doctorate in the field of Statistics, Mathematics or a related subject. They will be an outstanding individual who has the potential to become a leader in their field. The post holder will have the skills and enthusiasm to teach at undergraduate and graduate level, within the Department of Statistics, and to supervise student projects. They will carry out and publish original research within their area of specialisation. -

Genomics of Rare Disease 27-29 March 2019
Genomics of Rare Disease 27-29 March 2019 Wellcome Genome Campus, Hinxton, Cambridge, UK Conference Programme Wednesday 27 March 2019 12:00-13:30 Registration with lunch 13:30-13:40 Welcome and introduction Programme Committee: Matthew Hurles, Wellcome Sanger Institute, UK 13:40-14:40 Lupski lecture Chair: Matthew Hurles, Wellcome Sanger Institute, UK Exploring the “between” spaces: the continuum from mendelian to common disease, and the continuum from polygenic to multifactorial disease Nancy Cox Vanderbilt University Medical Center, USA 14:40-16:00 Session 1: Solving the unsolved Chair: Kym Boycott, Children's Hospital of Eastern Ontario – Research Institute, Canada 14:40 The NIH undiagnosed diseases program: expansion to a national and international networks William Gahl NIH, USA 15:20 Epigenetic dysregulation underpins tumourigenesis in a cutaneous tumour syndrome Neil Rajan Newcastle University, UK 15:35 Regulatory de novo mutations play significant role in severe intellectual disability Santosh Atanur Imperial College London, UK 15:50-16:30 Afternoon tea 1 16:30-17:00 Session 1 continued: Solving the unsolved Chair: Kym Boycott, Children's Hospital of Eastern Ontario – Research Institute, Canada 16:30 Exome sequencing for the diagnosis of ultrasound detected fetal abnormalities: a year’s experience at St George’s Hospital in partnership with Congenica Ltd Andrea Haworth Congenica Limited, UK 16:45 A bottom-up predictive approach to identify oligogenic disease causes Tom Lenaerts Université Libre de Bruxelles, Belgium 17:00-17:30 -

Nhbs Annual New and Forthcoming Titles Issue: 2003 Complete January 2004 [email protected] +44 (0)1803 865913
nhbs annual new and forthcoming titles Issue: 2003 complete January 2004 [email protected] +44 (0)1803 865913 The NHBS Monthly Catalogue in a complete yearly edition Zoology: Mammals Birds Welcome to the Complete 2003 edition of the NHBS Monthly Catalogue, the ultimate Reptiles & Amphibians buyer's guide to new and forthcoming titles in natural history, conservation and the Fishes environment. With 300-400 new titles sourced every month from publishers and research organisations around the world, the catalogue provides key bibliographic data Invertebrates plus convenient hyperlinks to more complete information and nhbs.com online Palaeontology shopping - an invaluable resource. Each month's catalogue is sent out as an HTML Marine & Freshwater Biology email to registered subscribers (a plain text version is available on request). It is also General Natural History available online, and offered as a PDF download. Regional & Travel Please see our info page for more details, also our standard terms and conditions. Botany & Plant Science Prices are correct at the time of publication, please check www.nhbs.com for the Animal & General Biology latest prices. NHBS Ltd, 2-3 Wills Rd, Totnes, Devon TQ9 5XN, UK Evolutionary Biology Ecology Habitats & Ecosystems Conservation & Biodiversity Environmental Science Physical Sciences Sustainable Development Data Analysis Reference Mammals An Affair with Red Squirrels 58 pages | Col photos | Larks Press David Stapleford Pbk | 2003 | 1904006108 | #143116A | Account of a lifelong passion, of the author's experience of breeding red squirrels, and more £5.00 BUY generally of their struggle for survival since the arrival of their grey .... All About Goats 178 pages | 30 photos | Whittet Lois Hetherington, J Matthews and LF Jenner Hbk | 2002 | 1873580606 | #138085A | A complete guide to keeping goats, including housing, feeding and breeding, rearing young, £15.99 BUY milking, dairy produce and by-products and showing. -
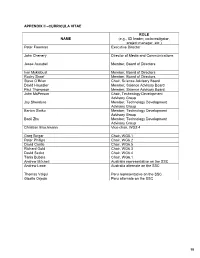
Appendix Ii –Curricula Vitae
APPENDIX II –CURRICULA VITAE ROL E NAME (e.g., ICI leader, co-investigator, project manager, etc.) Peter Freeman Executive Director John Chenery Director of Media and Communications Jesse Ausubel Member, Board of Directors Ivar Myklebust Member, Board of Directors Rocky Skeef Member, Board of Directors Steve O’Brien Chair, Science Advisory Board David Haussler Member, Science Advisory Board Paul Thompson Member, Science Advisory Board John McPerson Chair, Technology Development Advisory Group Jay Shendure Member, Technology Development Advisory Group Barton Slatko Member, Technology Development Advisory Group Baoli Zhu Member, Technology Development Advisory Group Christian Brochmann Vice-chair, WG2.4 Greg Singer Chair, WG5.1 Peter Phillips Chair, WG6.2 David Castle Chair, WG6.5 Richard Gold Chair, WG6.3 David Secko Chair, WG6.4 Tania Bubela Chair, WG6.1 Andrew Mitchell Australia representative on the SSC Andrew Lowe Australia alternate on the SSC Thomas Valqui Peru representative on the SSC Gisella Orjeda Peru alternate on the SSC 98 PETER FREEMAN BA, PHD, FIBD 120 Stuart Street, Guelph, Ontario, CANADA N1E 4S8 Phone (W): +1 519 824 4120 Cell: +1 519 731 2163 Email: [email protected] PROFILE Experienced science-based (PhD qualified) executive director, with a track record of successfully co- ordinating large multi-institutional research projects, networks and consortia in genomics, proteomics, stem cell research and population health. Strong financial management, strategic planning and project management skills gained in R&D and operations roles in the international malting and brewing industry Superior mentoring, facilitating, technical writing and editing skills used to develop successful interdisciplinary funding proposals. Fully proficient in information /communication technologies and their use in knowledge translation and public outreach activities.