Sustainable Software for Computational Chemistry and Materials Modeling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Free and Open Source Software for Computational Chemistry Education
Free and Open Source Software for Computational Chemistry Education Susi Lehtola∗,y and Antti J. Karttunenz yMolecular Sciences Software Institute, Blacksburg, Virginia 24061, United States zDepartment of Chemistry and Materials Science, Aalto University, Espoo, Finland E-mail: [email protected].fi Abstract Long in the making, computational chemistry for the masses [J. Chem. Educ. 1996, 73, 104] is finally here. We point out the existence of a variety of free and open source software (FOSS) packages for computational chemistry that offer a wide range of functionality all the way from approximate semiempirical calculations with tight- binding density functional theory to sophisticated ab initio wave function methods such as coupled-cluster theory, both for molecular and for solid-state systems. By their very definition, FOSS packages allow usage for whatever purpose by anyone, meaning they can also be used in industrial applications without limitation. Also, FOSS software has no limitations to redistribution in source or binary form, allowing their easy distribution and installation by third parties. Many FOSS scientific software packages are available as part of popular Linux distributions, and other package managers such as pip and conda. Combined with the remarkable increase in the power of personal devices—which rival that of the fastest supercomputers in the world of the 1990s—a decentralized model for teaching computational chemistry is now possible, enabling students to perform reasonable modeling on their own computing devices, in the bring your own device 1 (BYOD) scheme. In addition to the programs’ use for various applications, open access to the programs’ source code also enables comprehensive teaching strategies, as actual algorithms’ implementations can be used in teaching. -

The Molecular Sciences Software Institute
The Molecular Sciences Software Institute T. Daniel Crawford, Cecilia Clementi, Robert Harrison, Teresa Head-Gordon, Shantenu Jha*, Anna Krylov, Vijay Pande, and Theresa Windus http://molssi.org S2I2 HEP/CS Workshop at NCSA/UIUC 07 Dec, 2016 1 Outline • Space and Scope of Computational Molecular Sciences. • “State of the art and practice” • Intellectual drivers • Conceptualization Phase: Identifying the community and needs • Bio-molecular Simulations (BMS) Conceptualization • Quantum Mechanics/Chemistry (QM) Conceptualization • Execution Phase. • Structure and Governance Model • Resource Distribution • Work Plan 2 The Molecular Sciences Software Institute (MolSSI) • New project (as of August 1st, 2016) funded by the National Science Foundation. • Collaborative effort by Virginia Tech, Rice U., Stony Brook U., U.C. Berkeley, Stanford U., Rutgers U., U. Southern California, and Iowa State U. • Total budget of $19.42M for five years, potentially renewable to ten years. • Joint support from numerous NSF divisions: Advanced Cyberinfrastructure (ACI), Chemistry (CHE), Division of Materials Research (DMR), Office of Multidisciplinary Activities (OMA) • Designed to serve and enhance the software development efforts of the broad field of computational molecular science. 3 Computational Molecular Sciences (CMS) • The history of CMS – the sub-fields of quantum chemistry, computational materials science, and biomolecular simulation – reaches back decades to the genesis of computational science. • CMS is now a “full partner with experiment”. • For an impressive array of chemical, biochemical, and materials challenges, our community has developed simulations and models that directly impact: • Development of new chiral drugs; • Elucidation of the functionalities of biological macromolecules; • Development of more advanced materials for solar-energy storage, technology for CO2 sequestration, etc. -

Supporting Information
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information How to Select Ionic Liquids as Extracting Agent Systematically? Special Case Study for Extractive Denitrification Process Shurong Gaoa,b,c,*, Jiaxin Jina,b, Masroor Abroc, Ruozhen Songc, Miao Hed, Xiaochun Chenc,* a State Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing, 102206, China b Research Center of Engineering Thermophysics, North China Electric Power University, Beijing, 102206, China c Beijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China d Office of Laboratory Safety Administration, Beijing University of Technology, Beijing 100124, China * Corresponding author, Tel./Fax: +86-10-6443-3570, E-mail: [email protected], [email protected] 1 COSMO-RS Computation COSMOtherm allows for simple and efficient processing of large numbers of compounds, i.e., a database of molecular COSMO files; e.g. the COSMObase database. COSMObase is a database of molecular COSMO files available from COSMOlogic GmbH & Co KG. Currently COSMObase consists of over 2000 compounds including a large number of industrial solvents plus a wide variety of common organic compounds. All compounds in COSMObase are indexed by their Chemical Abstracts / Registry Number (CAS/RN), by a trivial name and additionally by their sum formula and molecular weight, allowing a simple identification of the compounds. We obtained the anions and cations of different ILs and the molecular structure of typical N-compounds directly from the COSMObase database in this manuscript. -

Open Babel Documentation Release 2.3.1
Open Babel Documentation Release 2.3.1 Geoffrey R Hutchison Chris Morley Craig James Chris Swain Hans De Winter Tim Vandermeersch Noel M O’Boyle (Ed.) December 05, 2011 Contents 1 Introduction 3 1.1 Goals of the Open Babel project ..................................... 3 1.2 Frequently Asked Questions ....................................... 4 1.3 Thanks .................................................. 7 2 Install Open Babel 9 2.1 Install a binary package ......................................... 9 2.2 Compiling Open Babel .......................................... 9 3 obabel and babel - Convert, Filter and Manipulate Chemical Data 17 3.1 Synopsis ................................................. 17 3.2 Options .................................................. 17 3.3 Examples ................................................. 19 3.4 Differences between babel and obabel .................................. 21 3.5 Format Options .............................................. 22 3.6 Append property values to the title .................................... 22 3.7 Filtering molecules from a multimolecule file .............................. 22 3.8 Substructure and similarity searching .................................. 25 3.9 Sorting molecules ............................................ 25 3.10 Remove duplicate molecules ....................................... 25 3.11 Aliases for chemical groups ....................................... 26 4 The Open Babel GUI 29 4.1 Basic operation .............................................. 29 4.2 Options ................................................. -

GROMACS: Fast, Flexible, and Free
GROMACS: Fast, Flexible, and Free DAVID VAN DER SPOEL,1 ERIK LINDAHL,2 BERK HESS,3 GERRIT GROENHOF,4 ALAN E. MARK,4 HERMAN J. C. BERENDSEN4 1Department of Cell and Molecular Biology, Uppsala University, Husargatan 3, Box 596, S-75124 Uppsala, Sweden 2Stockholm Bioinformatics Center, SCFAB, Stockholm University, SE-10691 Stockholm, Sweden 3Max-Planck Institut fu¨r Polymerforschung, Ackermannweg 10, D-55128 Mainz, Germany 4Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, NL-9747 AG Groningen, The Netherlands Received 12 February 2005; Accepted 18 March 2005 DOI 10.1002/jcc.20291 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: This article describes the software suite GROMACS (Groningen MAchine for Chemical Simulation) that was developed at the University of Groningen, The Netherlands, in the early 1990s. The software, written in ANSI C, originates from a parallel hardware project, and is well suited for parallelization on processor clusters. By careful optimization of neighbor searching and of inner loop performance, GROMACS is a very fast program for molecular dynamics simulation. It does not have a force field of its own, but is compatible with GROMOS, OPLS, AMBER, and ENCAD force fields. In addition, it can handle polarizable shell models and flexible constraints. The program is versatile, as force routines can be added by the user, tabulated functions can be specified, and analyses can be easily customized. Nonequilibrium dynamics and free energy determinations are incorporated. Interfaces with popular quantum-chemical packages (MOPAC, GAMES-UK, GAUSSIAN) are provided to perform mixed MM/QM simula- tions. The package includes about 100 utility and analysis programs. -

Chem Compute Quickstart
Chem Compute Quickstart Chem Compute is maintained by Mark Perri at Sonoma State University and hosted on Jetstream at Indiana University. The Chem Compute URL is https://chemcompute.org/. We will use Chem Compute as the frontend for running electronic structure calculations with The General Atomic and Molecular Electronic Structure System, GAMESS (http://www.msg.ameslab.gov/gamess/). Chem Compute also provides access to other computational chemistry resources including PSI4 and the molecular dynamics packages TINKER and NAMD, though we will not be using those resource at this time. Follow this link, https://chemcompute.org/gamess/submit, to directly access the Chem Compute GAMESS guided submission interface. If you are a returning Chem Computer user, please log in now. If your University is part of the InCommon Federation you can log in without registering by clicking Login then "Log In with Google or your University" – select your University from the dropdown list. Otherwise if this is your first time working with Chem Compute, please register as a Chem Compute user by clicking the “Register” link in the top-right corner of the page. This gives you access to all of the computational resources available at ChemCompute.org and will allow you to maintain copies of your calculations in your user “Dashboard” that you can refer to later. Registering also helps track usage and obtain the resources needed to continue providing its service. When logged in with the GAMESS-Submit tabs selected, an instruction section appears on the left side of the page with instructions for several different kinds of calculations. -

Computer-Assisted Catalyst Development Via Automated Modelling of Conformationally Complex Molecules
www.nature.com/scientificreports OPEN Computer‑assisted catalyst development via automated modelling of conformationally complex molecules: application to diphosphinoamine ligands Sibo Lin1*, Jenna C. Fromer2, Yagnaseni Ghosh1, Brian Hanna1, Mohamed Elanany3 & Wei Xu4 Simulation of conformationally complicated molecules requires multiple levels of theory to obtain accurate thermodynamics, requiring signifcant researcher time to implement. We automate this workfow using all open‑source code (XTBDFT) and apply it toward a practical challenge: diphosphinoamine (PNP) ligands used for ethylene tetramerization catalysis may isomerize (with deleterious efects) to iminobisphosphines (PPNs), and a computational method to evaluate PNP ligand candidates would save signifcant experimental efort. We use XTBDFT to calculate the thermodynamic stability of a wide range of conformationally complex PNP ligands against isomeriation to PPN (ΔGPPN), and establish a strong correlation between ΔGPPN and catalyst performance. Finally, we apply our method to screen novel PNP candidates, saving signifcant time by ruling out candidates with non‑trivial synthetic routes and poor expected catalytic performance. Quantum mechanical methods with high energy accuracy, such as density functional theory (DFT), can opti- mize molecular input structures to a nearby local minimum, but calculating accurate reaction thermodynamics requires fnding global minimum energy structures1,2. For simple molecules, expert intuition can identify a few minima to focus study on, but an alternative approach must be considered for more complex molecules or to eventually fulfl the dream of autonomous catalyst design 3,4: the potential energy surface must be frst surveyed with a computationally efcient method; then minima from this survey must be refned using slower, more accurate methods; fnally, for molecules possessing low-frequency vibrational modes, those modes need to be treated appropriately to obtain accurate thermodynamic energies 5–7. -

Living at the Top of the Top500: Myopia from Being at the Bleeding Edge
Living at the Top of the Top500: Myopia from Being at the Bleeding Edge Bronson Messer Oak Ridge Leadership Computing Facility & Theoretical Astrophysics Group Oak Ridge National Laboratory Department of Physics & Astronomy University of Tennessee Friday, July 1, 2011 Outline • Statements made without proof • OLCF’s Center for Accelerated Application Readiness • Speculations on task-based approaches for multiphysics applications in astrophysics (e.g. blowing up stars) 2 Friday, July 1, 2011 Riffing on Hank’s fable... 3 Friday, July 1, 2011 The Effects of Moore’s Law and Slacking 1 on Large Computations Chris Gottbrath, Jeremy Bailin, Casey Meakin, Todd Thompson, J.J. Charfman Steward Observatory, University of Arizona Abstract We show that, in the context of Moore’s Law, overall productivity can be increased for large enough computations by ‘slacking’orwaiting for some period of time before purchasing a computer and beginning the calculation. According to Moore’s Law, the computational power availableataparticular price doubles every 18 months. Therefore it is conceivable that for sufficiently large numerical calculations and fixed budgets, computing power will improve quickly enough that the calculation will finish faster if we wait until the available computing power is sufficiently better and start the calculation then. The Effects of Moore’s Law and Slacking 1Figureon Large 1: Computations Chris Gottbrath, Jeremy Bailin, Casey Meakin, Todd Thompson, J.J. Charfman 1 The Effects of Moore’sSteward Observatory, Law and University Slacking of Arizona on Large astro-ph/9912202 Computations Abstract Chris Gottbrath, Jeremy Bailin, Casey Meakin, Todd Thompson, We show that, in the context of Moore’s Law, overall productivity can be increased forJ.J. -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -

In Quantum Chemistry
http://www.cca-forum.org Computational Quality of Service (CQoS) in Quantum Chemistry Joseph Kenny1, Kevin Huck2, Li Li3, Lois Curfman McInnes3, Heather Netzloff4, Boyana Norris3, Meng-Shiou Wu4, Alexander Gaenko4 , and Hirotoshi Mori5 1Sandia National Laboratories, 2University of Oregon, 3Argonne National Laboratory, 4Ames Laboratory, 5Ochanomizu University, Japan This work is a collaboration among participants in the SciDAC Center for Technology for Advanced Scientific Component Software (TASCS), Performance Engineering Research Institute (PERI), Quantum Chemistry Science Application Partnership (QCSAP), and the Tuning and Analysis Utilities (TAU) group at the University of Oregon. Quantum Chemistry and the CQoS in Quantum Chemistry: Motivation and Approach Common Component Architecture (CCA) Motivation: CQoS Approach: CCA Overview: • QCSAP Challenges: How, during runtime, can we make the best choices • Overall: Develop infrastructure for dynamic component adaptivity, i.e., • The CCA Forum provides a specification and software tools for the for reliability, accuracy, and performance of interoperable quantum composing, substituting, and reconfiguring running CCA component development of high-performance components. chemistry components based on NWChem, MPQC, and GAMESS? applications in response to changing conditions – Performance, accuracy, mathematical consistency, reliability, etc. • Components = Composition – When several QC components provide the same functionality, what • Approach: Develop CQoS tools for – A component is a unit -

Parameterizing a Novel Residue
University of Illinois at Urbana-Champaign Luthey-Schulten Group, Department of Chemistry Theoretical and Computational Biophysics Group Computational Biophysics Workshop Parameterizing a Novel Residue Rommie Amaro Brijeet Dhaliwal Zaida Luthey-Schulten Current Editors: Christopher Mayne Po-Chao Wen February 2012 CONTENTS 2 Contents 1 Biological Background and Chemical Mechanism 4 2 HisH System Setup 7 3 Testing out your new residue 9 4 The CHARMM Force Field 12 5 Developing Topology and Parameter Files 13 5.1 An Introduction to a CHARMM Topology File . 13 5.2 An Introduction to a CHARMM Parameter File . 16 5.3 Assigning Initial Values for Unknown Parameters . 18 5.4 A Closer Look at Dihedral Parameters . 18 6 Parameter generation using SPARTAN (Optional) 20 7 Minimization with new parameters 32 CONTENTS 3 Introduction Molecular dynamics (MD) simulations are a powerful scientific tool used to study a wide variety of systems in atomic detail. From a standard protein simulation, to the use of steered molecular dynamics (SMD), to modelling DNA-protein interactions, there are many useful applications. With the advent of massively parallel simulation programs such as NAMD2, the limits of computational anal- ysis are being pushed even further. Inevitably there comes a time in any molecular modelling scientist’s career when the need to simulate an entirely new molecule or ligand arises. The tech- nique of determining new force field parameters to describe these novel system components therefore becomes an invaluable skill. Determining the correct sys- tem parameters to use in conjunction with the chosen force field is only one important aspect of the process. -
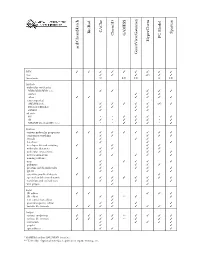
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only.