Characterization and Phylogenetic Analysis of The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

APPROVED PLANT LIST Midtown Alliance Tree Well Adoption Program
APPROVED PLANT LIST Midtown Alliance Tree Well Adoption Program Midtown Alliance launched the Tree Well Adoption program with the primary goal of enriching the experience of Midtown’s workers and residents while encouraging sustainability through the use of low-water, urban tolerant plant species. This list of plants was created to aid individuals and organizations in selecting plant material to plant in their adopted tree wells. This plant list is intended to encourage individual character in the tree wells, rather than restrict creativity in the selection of plants. The plants on the approved list were selected based on the following criteria: • Perennial. All plants listed are perennial, meaning they last for two or more growing seasons. Once established, these plants will require less water to maintain than annuals. • Heat tolerant. Plants in tree wells are exposed to high temperatures caused by vehicles and heat reflected from surrounding buildings, asphalt, and other urban surfaces. They must also be tolerant to high daytime temperatures, typical of Atlanta’s summer months, and cold hardy in the winter months. Atlanta is located in USDA Plant Hardiness Zone 7b/8a. • Water wise. Urban tree wells are surrounded by impervious surfaces and thus, are highly susceptible to periods of drought. Suitable plants must be able to survive periods of low rainfall. • Pollution tolerant. Vehicle exhaust may leave deposits and pollutants on plant foliage, which can kill sensitive plants. • Encourage wildlife. Flowering plants attract insects such as butterflies while others provide food sources for birds and other wildlife. • Grown locally. Many of the plants listed are native to the Atlanta area, and all can be found at local nurseries. -

Pdf (306.01 K)
REVIEW ARTICLE RECORDS OF PHARMACEUTICAL AND BIOMEDICAL SCIENCES Review article on chemical constituents and biological activity of Thymelaea hirsuta. Ahmed M Badawya, Hashem A Hassaneanb, Amany K. Ibrahimb, Eman S. Habibb, Safwat A. Ahmedb* aDepartment of Pharmacognosy, Faculty of Pharmacy, Sinai University, El-Arish, Egypt, b Department of Pharmacognosy, Faculty of Pharmacy, Suez Canal University, Ismailia, Egypt 41522. Abstract Received on: 07.04. 2019 Thymelaea hirsuta a perennial, evergreen and dioecious shrub, which is native Revised on: 30. 04. 2019 to North Africa. T. hirsuta is a widespread invasive weed and is commonly known as “Methnane”. Along the history, T. hirsuta, family Thymelaeaceae, Accepted on: 10. 04. 2019 has been recognized as an important medicinal plant. Much research has been carried out on the medical applications of Methnane. The choice of the plant was based on the good previous biological study of T. hirsuta plant extract to Correspondence Author: use as anticancer, hepatoprotective and anti-diapetic. Several species of Tel:+ 01092638387 Thymelaeaceae have been the subject of numerous phytochemical studies. Initially, interest may have been due to the marked toxicity of these plants, but E-mail address: the widespread use of some species medicinally has certainly played a part in [email protected] sustaining this interest. Keywords: Thymelaea hirsuta , Chemical constituents, Biological activity 1.Introduction: Near East: Lebanon and Palestine. The choice of the plant was based on the good previous Thymelaea hirsuta a perennial, evergreen and biological study of T. hirsuta plant extract to use dioecious shrub, which is native to North Africa. T. as anticancer, hepatoprotective and anti-diabetic. -

Thymelaeaceae)
Origin and diversification of the Australasian genera Pimelea and Thecanthes (Thymelaeaceae) by MOLEBOHENG CYNTHIA MOTS! Thesis submitted in fulfilment of the requirements for the degree PHILOSOPHIAE DOCTOR in BOTANY in the FACULTY OF SCIENCE at the UNIVERSITY OF JOHANNESBURG Supervisor: Dr Michelle van der Bank Co-supervisors: Dr Barbara L. Rye Dr Vincent Savolainen JUNE 2009 AFFIDAVIT: MASTER'S AND DOCTORAL STUDENTS TO WHOM IT MAY CONCERN This serves to confirm that I Moleboheng_Cynthia Motsi Full Name(s) and Surname ID Number 7808020422084 Student number 920108362 enrolled for the Qualification PhD Faculty _Science Herewith declare that my academic work is in line with the Plagiarism Policy of the University of Johannesburg which I am familiar. I further declare that the work presented in the thesis (minor dissertation/dissertation/thesis) is authentic and original unless clearly indicated otherwise and in such instances full reference to the source is acknowledged and I do not pretend to receive any credit for such acknowledged quotations, and that there is no copyright infringement in my work. I declare that no unethical research practices were used or material gained through dishonesty. I understand that plagiarism is a serious offence and that should I contravene the Plagiarism Policy notwithstanding signing this affidavit, I may be found guilty of a serious criminal offence (perjury) that would amongst other consequences compel the UJ to inform all other tertiary institutions of the offence and to issue a corresponding certificate of reprehensible academic conduct to whomever request such a certificate from the institution. Signed at _Johannesburg on this 31 of _July 2009 Signature Print name Moleboheng_Cynthia Motsi STAMP COMMISSIONER OF OATHS Affidavit certified by a Commissioner of Oaths This affidavit cordons with the requirements of the JUSTICES OF THE PEACE AND COMMISSIONERS OF OATHS ACT 16 OF 1963 and the applicable Regulations published in the GG GNR 1258 of 21 July 1972; GN 903 of 10 July 1998; GN 109 of 2 February 2001 as amended. -

Outline of Angiosperm Phylogeny
Outline of angiosperm phylogeny: orders, families, and representative genera with emphasis on Oregon native plants Priscilla Spears December 2013 The following listing gives an introduction to the phylogenetic classification of the flowering plants that has emerged in recent decades, and which is based on nucleic acid sequences as well as morphological and developmental data. This listing emphasizes temperate families of the Northern Hemisphere and is meant as an overview with examples of Oregon native plants. It includes many exotic genera that are grown in Oregon as ornamentals plus other plants of interest worldwide. The genera that are Oregon natives are printed in a blue font. Genera that are exotics are shown in black, however genera in blue may also contain non-native species. Names separated by a slash are alternatives or else the nomenclature is in flux. When several genera have the same common name, the names are separated by commas. The order of the family names is from the linear listing of families in the APG III report. For further information, see the references on the last page. Basal Angiosperms (ANITA grade) Amborellales Amborellaceae, sole family, the earliest branch of flowering plants, a shrub native to New Caledonia – Amborella Nymphaeales Hydatellaceae – aquatics from Australasia, previously classified as a grass Cabombaceae (water shield – Brasenia, fanwort – Cabomba) Nymphaeaceae (water lilies – Nymphaea; pond lilies – Nuphar) Austrobaileyales Schisandraceae (wild sarsaparilla, star vine – Schisandra; Japanese -
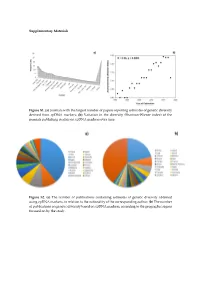
(A) Journals with the Largest Number of Papers Reporting Estimates Of
Supplementary Materials Figure S1. (a) Journals with the largest number of papers reporting estimates of genetic diversity derived from cpDNA markers; (b) Variation in the diversity (Shannon-Wiener index) of the journals publishing studies on cpDNA markers over time. Figure S2. (a) The number of publications containing estimates of genetic diversity obtained using cpDNA markers, in relation to the nationality of the corresponding author; (b) The number of publications on genetic diversity based on cpDNA markers, according to the geographic region focused on by the study. Figure S3. Classification of the angiosperm species investigated in the papers that analyzed genetic diversity using cpDNA markers: (a) Life mode; (b) Habitat specialization; (c) Geographic distribution; (d) Reproductive cycle; (e) Type of flower, and (f) Type of pollinator. Table S1. Plant species identified in the publications containing estimates of genetic diversity obtained from the use of cpDNA sequences as molecular markers. Group Family Species Algae Gigartinaceae Mazzaella laminarioides Angiospermae Typhaceae Typha laxmannii Angiospermae Typhaceae Typha orientalis Angiospermae Typhaceae Typha angustifolia Angiospermae Typhaceae Typha latifolia Angiospermae Araliaceae Eleutherococcus sessiliflowerus Angiospermae Polygonaceae Atraphaxis bracteata Angiospermae Plumbaginaceae Armeria pungens Angiospermae Aristolochiaceae Aristolochia kaempferi Angiospermae Polygonaceae Atraphaxis compacta Angiospermae Apocynaceae Lagochilus macrodontus Angiospermae Polygonaceae Atraphaxis -

Leafing Through History
Leafing Through History Leafing Through History Several divisions of the Missouri Botanical Garden shared their expertise and collections for this exhibition: the William L. Brown Center, the Herbarium, the EarthWays Center, Horticulture and the William T. Kemper Center for Home Gardening, Education and Tower Grove House, and the Peter H. Raven Library. Grateful thanks to Nancy and Kenneth Kranzberg for their support of the exhibition and this publication. Special acknowledgments to lenders and collaborators James Lucas, Michael Powell, Megan Singleton, Mimi Phelan of Midland Paper, Packaging + Supplies, Dr. Shirley Graham, Greg Johnson of Johnson Paper, and the Campbell House Museum for their contributions to the exhibition. Many thanks to the artists who have shared their work with the exhibition. Especial thanks to Virginia Harold for the photography and Studiopowell for the design of this publication. This publication was printed by Advertisers Printing, one of only 50 U.S. printing companies to have earned SGP (Sustainability Green Partner) Certification, the industry standard for sustainability performance. Copyright © 2019 Missouri Botanical Garden 2 James Lucas Michael Powell Megan Singleton with Beth Johnson Shuki Kato Robert Lang Cekouat Léon Catherine Liu Isabella Myers Shoko Nakamura Nguyen Quyet Tien Jon Tucker Rob Snyder Curated by Nezka Pfeifer Museum Curator Stephen and Peter Sachs Museum Missouri Botanical Garden Inside Cover: Acapulco Gold rolling papers Hemp paper 1972 Collection of the William L. Brown Center [WLBC00199] Previous Page: Bactrian Camel James Lucas 2017 Courtesy of the artist Evans Gallery Installation view 4 Plants comprise 90% of what we use or make on a daily basis, and yet, we overlook them or take them for granted regularly. -

Heterodichogamy.Pdf
Research Update TRENDS in Ecology & Evolution Vol.16 No.11 November 2001 595 How common is heterodichogamy? Susanne S. Renner The sexual systems of plants usually Heterodichogamy differs from normal (Zingiberales). These figures probably depend on the exact spatial distribution of dichogamy, the temporal separation of underestimate the frequency of the gamete-producing structures. Less well male and female function in flowers, in heterodichogamy. First, the phenomenon known is how the exact timing of male and that it involves two genetic morphs that is discovered only if flower behavior is female function might influence plant occur at a 1:1 ratio. The phenomenon was studied in several individuals and in mating. New papers by Li et al. on a group discovered in walnuts and hazelnuts5,6 natural populations. Differential of tropical gingers describe differential (the latter ending a series of Letters to movements and maturation of petals, maturing of male and female structures, the Editor about hazel flowering that styles, stigmas and stamens become such that half the individuals of a began in Nature in 1870), but has gone invisible in dried herbarium material, population are in the female stage when almost unnoticed7. Indeed, its recent and planted populations deriving from the other half is in the male stage. This discovery in Alpinia was greeted as a vegetatively propagated material no new case of heterodichogamy is unique new mechanism, differing ‘from other longer reflect natural morph ratios. The in involving reciprocal movement of the passive outbreeding devices, such as discovery of heterodichogamy thus styles in the two temporal morphs. dichogamy…and heterostyly in that it depends on field observations. -

Guidance Document Pohakuloa Training Area Plant Guide
GUIDANCE DOCUMENT Recovery of Native Plant Communities and Ecological Processes Following Removal of Non-native, Invasive Ungulates from Pacific Island Forests Pohakuloa Training Area Plant Guide SERDP Project RC-2433 JULY 2018 Creighton Litton Rebecca Cole University of Hawaii at Manoa Distribution Statement A Page Intentionally Left Blank This report was prepared under contract to the Department of Defense Strategic Environmental Research and Development Program (SERDP). The publication of this report does not indicate endorsement by the Department of Defense, nor should the contents be construed as reflecting the official policy or position of the Department of Defense. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the Department of Defense. Page Intentionally Left Blank 47 Page Intentionally Left Blank 1. Ferns & Fern Allies Order: Polypodiales Family: Aspleniaceae (Spleenworts) Asplenium peruvianum var. insulare – fragile fern (Endangered) Delicate ENDEMIC plants usually growing in cracks or caves; largest pinnae usually <6mm long, tips blunt, uniform in shape, shallowly lobed, 2-5 lobes on acroscopic side. Fewer than 5 sori per pinna. Fronds with distal stipes, proximal rachises ocassionally proliferous . d b a Asplenium trichomanes subsp. densum – ‘oāli’i; maidenhair spleenwort Plants small, commonly growing in full sunlight. Rhizomes short, erect, retaining many dark brown, shiny old stipe bases.. Stipes wiry, dark brown – black, up to 10cm, shiny, glabrous, adaxial surface flat, with 2 greenish ridges on either side. Pinnae 15-45 pairs, almost sessile, alternate, ovate to round, basal pinnae smaller and more widely spaced. -

Stellera Chamaejasme L
Annals of Microbiology (2018) 68:273–286 https://doi.org/10.1007/s13213-018-1336-0 ORIGINAL ARTICLE Bacterial community structure associated with the rhizosphere soils and roots of Stellera chamaejasme L. along a Tibetan elevation gradient Hui Jin1,2 & Xiaoyan Yang2 & Rentao Liu1 & Zhiqiang Yan2 & Xudong Li3 & Xiuzhuang Li2 & Anxiang Su4 & Yuhui Zhao5 & Bo Qin2 Received: 27 November 2017 /Accepted: 22 March 2018 /Published online: 30 March 2018 # Springer-Verlag GmbH Germany, part of Springer Nature and the University of Milan 2018 Abstract The effect of altitude on the composition and diversity of microbial communities have attracted highly attention recently but is still poorly understood. We used 16S rRNA gene clone library analyses to characterize the bacterial communities from the rhizosphere and roots of Stellera chamaejasme in the Tibetan Plateau. Our results revealed that Actinobacteria and Proteobacteria were dominant bacteria in this medicinal plant in the rhizosphere and root communities. The Shannon diversity index showed that the bacterial diversity of rhizosphere follows a small saddle pattern, while the roots possesses of a hump-backed trend. Significant differences in the composition of bacterial communities between rhizosphere and roots were detected based on multiple com- parisons analysis. The community of Actinobacteria was found to be significantly negative correlated with soil available P (p < 0.01), while the phylum of Proteobacteria showed a positive relationship with available P (p < 0.05). Moreover, redundancy analysis indicated that soil phosphorus, pH, latitude, elevation and potassium positively correlated with bacterial communities associated with rhizosphere soils. Taken together, we provide evidence that bacterial communities associated with S. -

Biodiversity of the Hypersaline Urmia Lake National Park (NW Iran)
Diversity 2014, 6, 102-132; doi:10.3390/d6010102 OPEN ACCESS diversity ISSN 1424-2818 www.mdpi.com/journal/diversity Review Biodiversity of the Hypersaline Urmia Lake National Park (NW Iran) Alireza Asem 1,†,*, Amin Eimanifar 2,†,*, Morteza Djamali 3, Patricio De los Rios 4 and Michael Wink 2 1 Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao 266003, China 2 Institute of Pharmacy and Molecular Biotechnology (IPMB), Heidelberg University, Im Neuenheimer Feld 364, Heidelberg D-69120, Germany; E-Mail: [email protected] 3 Institut Méditerranéen d’Ecologie et de Paléoécologie UMR 6116 du CNRS-Europôle Méditerranéen de l’Arbois-Pavillon Villemin-BP 80, Aix-en-Provence Cedex 04 13545, France; E-Mail: [email protected] 4 Environmental Sciences School, Natural Resources Faculty, Catholic University of Temuco, Casilla 15-D, Temuco 4780000, Chile; E-Mail: [email protected] † These authors contributed equally to this work. * Authors to whom correspondence should be addressed; E-Mails: [email protected] (A.A.); [email protected] (A.E.); Tel.: +86-150-6624-4312 (A.A.); Fax: +86-532-8203-2216 (A.A.); Tel.: +49-6221-544-880 (A.E.); Fax: +49-6221-544-884 (A.E.). Received: 3 December 2013; in revised form: 13 January 2014 / Accepted: 27 January 2014 / Published: 10 February 2014 Abstract: Urmia Lake, with a surface area between 4000 to 6000 km2, is a hypersaline lake located in northwest Iran. It is the saltiest large lake in the world that supports life. Urmia Lake National Park is the home of an almost endemic crustacean species known as the brine shrimp, Artemia urmiana. -

Vascular Plant Families of the United States Grouped by Diagnostic Features
Humboldt State University Digital Commons @ Humboldt State University Botanical Studies Open Educational Resources and Data 12-6-2019 Vascular Plant Families of the United States Grouped by Diagnostic Features James P. Smith Jr Humboldt State University, [email protected] Follow this and additional works at: https://digitalcommons.humboldt.edu/botany_jps Part of the Botany Commons Recommended Citation Smith, James P. Jr, "Vascular Plant Families of the United States Grouped by Diagnostic Features" (2019). Botanical Studies. 96. https://digitalcommons.humboldt.edu/botany_jps/96 This Flora of the United States and North America is brought to you for free and open access by the Open Educational Resources and Data at Digital Commons @ Humboldt State University. It has been accepted for inclusion in Botanical Studies by an authorized administrator of Digital Commons @ Humboldt State University. For more information, please contact [email protected]. FLOWERING PLANT FAMILIES OF THE UNITED STATES GROUPED BY DIAGNOSTIC FEATURES James P. Smith, Jr. Professor Emeritus of Botany Department of Biological Sciences Humboldt State University Second edition — 6 December 2019 The focus is on families of plants found in the conterminous United States, including ornamentals. The listing of a family is not meant to imply that every species has that feature. I am using a fewfamily names, such as Liliaceae, Plantaginaceae, and Scrophulariaceae, in the traditional sense, because their limits remain unsettled. Parasitic on branches Dioscoreaceae -

Illustrated Flora of East Texas Illustrated Flora of East Texas
ILLUSTRATED FLORA OF EAST TEXAS ILLUSTRATED FLORA OF EAST TEXAS IS PUBLISHED WITH THE SUPPORT OF: MAJOR BENEFACTORS: DAVID GIBSON AND WILL CRENSHAW DISCOVERY FUND U.S. FISH AND WILDLIFE FOUNDATION (NATIONAL PARK SERVICE, USDA FOREST SERVICE) TEXAS PARKS AND WILDLIFE DEPARTMENT SCOTT AND STUART GENTLING BENEFACTORS: NEW DOROTHEA L. LEONHARDT FOUNDATION (ANDREA C. HARKINS) TEMPLE-INLAND FOUNDATION SUMMERLEE FOUNDATION AMON G. CARTER FOUNDATION ROBERT J. O’KENNON PEG & BEN KEITH DORA & GORDON SYLVESTER DAVID & SUE NIVENS NATIVE PLANT SOCIETY OF TEXAS DAVID & MARGARET BAMBERGER GORDON MAY & KAREN WILLIAMSON JACOB & TERESE HERSHEY FOUNDATION INSTITUTIONAL SUPPORT: AUSTIN COLLEGE BOTANICAL RESEARCH INSTITUTE OF TEXAS SID RICHARDSON CAREER DEVELOPMENT FUND OF AUSTIN COLLEGE II OTHER CONTRIBUTORS: ALLDREDGE, LINDA & JACK HOLLEMAN, W.B. PETRUS, ELAINE J. BATTERBAE, SUSAN ROBERTS HOLT, JEAN & DUNCAN PRITCHETT, MARY H. BECK, NELL HUBER, MARY MAUD PRICE, DIANE BECKELMAN, SARA HUDSON, JIM & YONIE PRUESS, WARREN W. BENDER, LYNNE HULTMARK, GORDON & SARAH ROACH, ELIZABETH M. & ALLEN BIBB, NATHAN & BETTIE HUSTON, MELIA ROEBUCK, RICK & VICKI BOSWORTH, TONY JACOBS, BONNIE & LOUIS ROGNLIE, GLORIA & ERIC BOTTONE, LAURA BURKS JAMES, ROI & DEANNA ROUSH, LUCY BROWN, LARRY E. JEFFORDS, RUSSELL M. ROWE, BRIAN BRUSER, III, MR. & MRS. HENRY JOHN, SUE & PHIL ROZELL, JIMMY BURT, HELEN W. JONES, MARY LOU SANDLIN, MIKE CAMPBELL, KATHERINE & CHARLES KAHLE, GAIL SANDLIN, MR. & MRS. WILLIAM CARR, WILLIAM R. KARGES, JOANN SATTERWHITE, BEN CLARY, KAREN KEITH, ELIZABETH & ERIC SCHOENFELD, CARL COCHRAN, JOYCE LANEY, ELEANOR W. SCHULTZE, BETTY DAHLBERG, WALTER G. LAUGHLIN, DR. JAMES E. SCHULZE, PETER & HELEN DALLAS CHAPTER-NPSOT LECHE, BEVERLY SENNHAUSER, KELLY S. DAMEWOOD, LOGAN & ELEANOR LEWIS, PATRICIA SERLING, STEVEN DAMUTH, STEVEN LIGGIO, JOE SHANNON, LEILA HOUSEMAN DAVIS, ELLEN D.