Human DECR1 Is an Androgen-Repressed Survival Factor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Low Resolution
Australian Biochemist The Magazine of the Australian Society for Biochemistry and Molecular Biology Inc. August 2019, Volume 50, Number 2 VOLISSN 50 NO 2 AUGUST 1443-0193 2019 AUSTRALIAN BIOCHEMIST PAGE 1 Be inspired during the 3- da y progra m including a fa nta stic lineup of industry lea ding interna tiona l plena ry spea kers, a nd hea r from our society specia lty lectures, be enga ged with poster presenta tions a nd lea rn a t the E/MCR mini- symposium. For more informa tion on our spea kers, progra m a nd to register, visit our website www.a sbmb2019.com.a u REGISTER NOW! PAGE 2 AUSTRALIAN BIOCHEMIST VOL 50 NO 2 AUGUST 2019 Table of Contents 4 Editorial Committee 5 Editorial 7 Publications with Impact Distinct Mechanisms Govern Recognition of Viral and Host Ligands by an Innate Immune Receptor Buying Time for Contractile Signaling Genetic Stutters, Gut Feelings and Neurodegenerative Disease 11 Off the Beaten Track Why it Sometimes Pays to Work for Money 14 ASBMB Education Feature Glycogen Builder: the Game Making a Drama Out of Biochemistry From the Whiteboard to the Conference: Scaffolding Conference-style Poster Presentations 18 SDS Page The Importance of Blue Sky Research 19 Competition: Unscramble 20 ASBMB 2019 International Plenary Speaker Profiles 22 ASBMB 2019 Symposium Speakers 23 Melbourne Protein Group: an ASBMB Special Interest Group 24 ASBMB Shimadzu Education Award Report 26 Intellectual Property Patenting Inventions in the Microbiome Space 29 Queen’s Birthday Honours for ASBMB Members 31 ASBMB Awards 2020 33 Election of Council 2020 33 Annual General Meeting of ASBMB 34 New ASBMB Members 35 Forthcoming Meetings 36 Our Sustaining Members 41 ASBMB Council 42 Directory Front Cover Bachelor of Biomedicine students from the University of Melbourne participating in an acting skills workshop with Rinske Ginsberg, VCA Theatre, as part of the Performing Sciences program, where students devise short performances embodying biochemical concepts. -

Pfcerli1 Is a Conserved Rhoptry Associated Protein Essential for Plasmodium Falciparum Merozoite Invasion of Erythrocytes
ARTICLE https://doi.org/10.1038/s41467-020-15127-w OPEN PfCERLI1 is a conserved rhoptry associated protein essential for Plasmodium falciparum merozoite invasion of erythrocytes Benjamin Liffner 1,8, Sonja Frölich 1,8, Gary K. Heinemann2, Boyin Liu3, Stuart A. Ralph 3, ✉ Matthew W.A. Dixon 3, Tim-Wolf Gilberger4,5,6 & Danny W. Wilson 1,7 1234567890():,; The disease-causing blood-stage of the Plasmodium falciparum lifecycle begins with invasion of human erythrocytes by merozoites. Many vaccine candidates with key roles in binding to the erythrocyte surface and entry are secreted from the large bulb-like rhoptry organelles at the apical tip of the merozoite. Here we identify an essential role for the conserved protein P. falciparum Cytosolically Exposed Rhoptry Leaflet Interacting protein 1 (PfCERLI1) in rhoptry function. We show that PfCERLI1 localises to the cytosolic face of the rhoptry bulb membrane and knockdown of PfCERLI1 inhibits merozoite invasion. While schizogony and merozoite organelle biogenesis appear normal, biochemical techniques and semi-quantitative super- resolution microscopy show that PfCERLI1 knockdown prevents secretion of key rhoptry antigens that coordinate merozoite invasion. PfCERLI1 is a rhoptry associated protein iden- tified to have a direct role in function of this essential merozoite invasion organelle, which has broader implications for understanding apicomplexan invasion biology. 1 Research Centre for Infectious Diseases, School of Biological Sciences, University of Adelaide, Adelaide, SA 5005, Australia. 2 Experimental Therapeutics Laboratory, School of Pharmacy and Medical Sciences, University of South Australia Cancer Research Institute, Adelaide, SA 5005, Australia. 3 Department of Biochemistry and Molecular Biology, Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Melbourne, VIC 3010, Australia. -

Melbourne Biomedical Precinct Studley Rd to an Exceptional Network Economic and Investment Growth Swinburne University of of Skilled Workers, Quality for the State
The Royal Children’s Hospital + The Murdoch Children’s Research Institute The Royal Women’s Hospital Darwin Walter & Eliza Hall Institute Monash Institute of Pharmaceutical NT Science, Monash University + CSIRO QLD Royal Melbourne Hospital WA Brisbane C Florey Institute SA e m ete ry Rd The University of Melbourne NSW Perth Sydney A Precinct approach Victorian Comprehensive Cancer Centre Adelaide Royal Pde Canberra + Peter MacCallum Cancer Centre The Doherty Institute to collaboration Flemington Rd Elgin St St Vincent’s Hospital Melbourne Hobart Melbourne, Grattan St Swanston St Australia and innovation Arden St Curzon St Elizabeth St Lygon St La Trobe University Queensberry St d R g r e b Burgundy St Rathdowne St l Chetwynd St Victoria St e > id e Melbourne Brain Centre H r Peel St e Austin Health p Plan Melbourne, Victoria’s key metropolitan Franklin St p Melbourne has biomedical La Trobe St U Austin Hospital planning strategy, highlights the precincts Banksia St capabilities unparalleled in around Melbourne, Monash, Deakin, Olivia Newton-John Australia and amongst the and La Trobe universities as clusters CSL Cancer Wellness & world’s best. We are home which offer significant opportunities (Poplar Road) Research Centre for innovation as well as employment, Melbourne Biomedical Precinct Studley Rd to an exceptional network economic and investment growth Swinburne University of of skilled workers, quality for the state. Technology education providers, leading The Melbourne Biomedical Precinct Deakin University St Kilda Rd research institutes and a Partners are largely collocated to the Princes Hwy Commercial Rd Burwood & Geelong Campuses sophisticated health system. north of Melbourne’s CBD close to Monash University, Australia’s highest ranking university, Clayton � Caulfield There is a growing body of evidence the University of Melbourne. -

University of Melbourne Annualreport2017 Fullreport
Annual Report 2017 unimelb.edu.au Contents Chancellor’s letter 1 VC’s introduction 2 The Melbourne Vision 3 Our past, present and future 4 2017 timeline 6 At a glance 8 Five-year statistics 10 Growing Esteem 12 Chapters in brief 14 Teaching, Learning and the Student Experience 16 Research 34 Engagement 48 Sustainability 64 Staff honours 78 High-achieving students 80 University governance 82 Council members 83 Academic governance 86 Governance structure 87 Senior leadership/University management 88 Statutory reporting 92 Financial report 105 Financial report index 106 Financial statement overview 107 Five-year financial summary 110 Financial statements 114 Disclosure index 166 Glossary 169 Index 171 | Front cover: ‘Eagle nest’, 2011, acrylic on canvas by Wurundjeri/Yorta Yorta artist and researcher, Ashley Kerr-Firebrace, is an intricate depiction of Wurundjeri creator Bunjil (the eagle) standing strong in the nest and supported by country, culture and people. The University of Melbourne stands on the land of the Wurundjeri people of the Kulin Nations. Ashley is the son of Wurundjeri Elder, Aunty Diane Kerr, who was born in Carlton, lives on Country and performs the Wominjeka (Welcome) to Country at University of Melbourne events and ceremonies. The Hon. Gayle Tierney MLC Minister for Training and Skills Level 1, 2 Treasury Place East Melbourne Vic 3002 16 March 2018 Dear Minister In accordance with the requirements of regulations and financial reporting directions under the Financial Management Act 1994, I am pleased to submit for your information and presentation to Parliament the Annual Report of the University of Melbourne for the year ending 31 December 2017. -

DECR1 Is an Androgen-Repressed Survival Factor That Regulates PUFA Oxidation To
bioRxiv preprint doi: https://doi.org/10.1101/865626; this version posted December 6, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis Zeyad D. Nassar1,2*, Chui Yan Mah1,2*, Jonas Dehairs3, Ingrid J.G. Burvenich4, Swati Irani1,2, Margaret M. Centenera1,2, Raj K. Shrestha5, Max Moldovan2, Anthony S. Don6, Andrew M. Scott4, Lisa G. Horvath7, David J. Lynn2,8, Luke A. Selth1,5,8, Andrew J. Hoy9, Johannes V. Swinnen3, Lisa M. Butler1,2# 1 University of Adelaide Medical School and Freemasons Foundation Centre for Men’s Health, University of Adelaide, Adelaide, SA 5005, Australia 2 South Australian Health and Medical Research Institute, Adelaide, SA 5000, Australia 3 KU Leuven- University of Leuven, LKI- Leuven Cancer Institute, Department of Oncology, Laboratory of Lipid Metabolism and Cancer, Leuven, B-3000, Belgium 4 Tumour Targeting Laboratory, Olivia Newton-John Cancer Research Institute, and School of Cancer Medicine, La Trobe University, Melbourne, VIC 3084, Australia 5 Dame Roma Mitchell Cancer Research Laboratories, University of Adelaide, Adelaide, SA 5005, Australia 6 NHMRC Clinical Trials Centre, and Centenary Institute, The University of Sydney, Camperdown, NSW 2006, Australia 7 Garvan Institute of Medical Research, Darlinghurst, NSW 2010; University of Sydney, Camperdown, NSW 2006; and University of New South Wales, Sydney, NSW 2052, Australia 8 College of Medicine and Public Health, Flinders University, Bedford Park, SA 5042, Australia 1 bioRxiv preprint doi: https://doi.org/10.1101/865626; this version posted December 6, 2019. -

CSL Research - Investor Site Tour @ Bio21 Institute 30 April 2018
CSL Research - Investor Site Tour @ Bio21 Institute 30 April 2018 CSL Research • Bio21 / Parkville, Marburg, Bern • Protein, gene & cell based therapies Specialty Breakthrough Immunoglobulins Haemophilia Transplantation Products Medicines • ~200 staff • Molecular Biology, Protein Biochemistry, Cell Biology & Physiology, Bioinformatics, Research & Clinical Analytics, Pharm/Tox Plasma Protein Science & Discovery 2 | Driven by Our Promise™ Global Hub for Research & Translational Medicine • New laboratories being constructed on the Bio21 site, 3628m2 • CSL space from 1470m2 to 3500m2 • Capacity to increase from 75 FTE’s to 165 (relocation from PKV) • University of Melbourne to fund construction • CSL to fund fit-out of CSL space • Construction commenced Q3 2016 with fit-out completed Q3 2018 3 | Driven by Our Promise™ Melbourne / Parkville Biomedical Precinct • a CSL competitive advantage Bio21 CSL 4 | Driven by Our Promise™ Melbourne / Parkville Biomedical Precinct • a CSL competitive advantage Melb. University Royal Melb. Florey Inst. Medical School Hospital Neuroscience Doherty Inst. Immunol. Microbiol Aust. Genomics Research Facility Victorian Comprehensive Cancer Centre Peter Mac WEHI Bio21 Cancer Inst. Women’s Hospital Royal Children's Murdoch Research Hospital Institute 5 | Driven by Our Promise™ WEHI / CSL Bioinformatics Alliance WEHI - world leaders in bioinformatic software development • By research specialty - differential expression - regulation of gene expression - cancer genome analysis - statistical genetics - molecular epidemiology -

The Bio21 Institute's 2020 Annual Report Is Available to Download
Annual Report 2020 Image of the coronavirus SARS-CoV-2 taken by Andrew Leis and Jason Roberts. Courtesy of the Doherty Institute. The Bio21 Molecular Science and Director Associate Director – Platform Biotechnology Institute Professor Michael W. Parker Infrastructure University of Melbourne DPhil (Oxon) FAA FAHMS Professor Malcolm McConville PhD 30 Flemington Road Deputy Director Associate Director – Commercialisation Parkville Victoria 3010 Professor Frances Separovic AO Professor Spencer Williams PhD Telephone: (03) 8344 2220 PhD FAA www.bio21.unimelb.edu.au Associate Director – Engagement @Bio21Institute Professor Sally Gras PhD @Bio21Institute Scientific Research Manager Dr David Keizer, PhD Produced by the Bio21 Molecular Science and Biotechnology External Relations Advisor,b Bio21 Florienne Institute Loder Annual Report 2020 Contents Our Mission 2 Our Vision 2 About the Institute 3 Director’s Message 4 Bio21 Leadership 8 Deputy Director, Professor Emeritus Frances Separovic AO 8 Associate Director Engagement – Professor Sally Gras 10 Associate Director Commercialisation – Professor Spencer Williams 13 Associate Director Platform Infrastructure – Professor Malcolm McConville 14 ACRF Facility for Innovative Cancer Drug Discovery 16 Impacts of Research 19 OHS Report 31 Equity Diversity and Inclusion 32 Industry Engagement and Commercialisation 35 Announcing the Ruth Bishop Building and Ian Holmes Imaging Centre 36 Events and Conferences 39 Graduate Research Students and Early Career Researchers 41 Institute Members Honoured 42 Grant Successes 43 Governance 46 Bio21 Scientific Research Team 47 Bio21 Research Groups 48 Bio21 People 50 Institute in Numbers 56 Bio21 Institute Theses submitted in 2020 57 Bio21 Steering Committee 58 Industry partners 63 Produced by the Bio21 Molecular Science aFontn Front cover image: Bio21 precinct aerial photograph, courtesy of Kane Jarrod Photography. -
2Nd Postdoc Methods Symposium Program
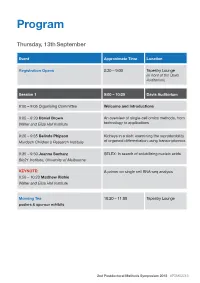
Program Thursday, 13th September Event Approximate Time Location Registration Opens 8:30 – 9:00 Tapestry Lounge (in front of the Davis Auditorium) Session 1 9:00 – 10:20 Davis Auditorium 9:00 – 9:05 Organising Committee Welcome and Introductions 9:05 – 9:20 Daniel Brown An overview of single-cell omics methods, from Walter and Eliza Hall Institute technology to applications 9:20 – 9:35 Belinda Phipson Kidneys in a dish: examining the reproducibility Murdoch Children’s Research Institute of organoid differentiation using transcriptomics 9:35 – 9:50 Joanna Sacharz SELEX: In search of solubilizing nucleic acids Bio21 Institute, University of Melbourne KEYNOTE: A primer on single cell RNA-seq analysis 9:50 – 10:20 Matthew Richie Walter and Eliza Hall Institute Morning Tea 10:20 – 11:00 Tapestry Lounge posters & sponsor exhibits 2nd Postdoctoral Methods Symposium 2018 #PDMS2018 Event Approximate Time Location Session 2 11:00 – 12:15 Davis Auditorium MCFP sponsored talk Biological Imaging via Helium ion Microscopy 11:00 – 11:15 Babak Nasr University of Melbourne 11:15 – 11:30 Mohamed Fareh Single-molecule fluorescence reveals the Peter MacCallum Cancer Centre dynamics of microRNA recognition by Dicer- TRBP complex 11:30 – 11:45 Charis Teh Capturing the cellular gymnastics of survival Walter and Eliza Hall institute and killer proteins by mass cytometry (CyTOF) 11:45 – 12:00 Jieqiong Lou Phasor analysis and image correlation Bio21 Institute, University of Melbourne spectroscopy of histone FLIM/FRET reveals spatiotemporal regulation of chromatin organization by the DNA damage response. Session 2 Flash talks 12:00 – 12:15 Davis Auditorium Rachel Lundie Flow FISH as a method to elucidate the Walter and Eliza Hall Institute transcriptional effects of M. -

Evolutionary Analyses of the Major Variant Surface Antigen-Encoding Genes Reveal Population Structure of Plasmodium Falciparum Within and Between Continents
RESEARCH ARTICLE Evolutionary analyses of the major variant surface antigen-encoding genes reveal population structure of Plasmodium falciparum within and between continents 1,2,3 1¤ 1,4 Gerry Tonkin-HillID , Shazia Ruybal-PesaÂntezID , Kathryn E. TiedjeID , 5 1,4 6 7,8 Virginie RougeronID , Michael F. DuffyID , Sedigheh Zakeri , Tepanata PumpaiboolID , 8,9 10,11 11 Pongchai HarnyuttanakornID , OraLee H. BranchID , Lastenia Ruiz-MesõÂaID , 1 5 2,12,13,14,15 a1111111111 Thomas S. Rask , Franck PrugnolleID , Anthony T. PapenfussID , Yao- 12,16 1,4 a1111111111 ban ChanID , Karen P. DayID * a1111111111 1 School of BioSciences, Bio21 Institute, The University of Melbourne, Melbourne, Australia, a1111111111 2 Bioinformatics Division, Walter and Eliza Hall Institute, Melbourne, Australia, 3 Parasites and Microbes, a1111111111 Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton, United Kingdom, 4 Department of Microbiology and Immunology, Bio21 Institute and Peter Doherty Institute, The University of Melbourne, Melbourne, Australia, 5 Laboratoire MIVEGEC, Universite de Montpellier-CNRS-IRD, Montpellier, France, 6 Malaria and Vector Research Group (MVRG), Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran, 7 Biomedical Science, Graduate School, Chulalongkorn University, Bangkok, Thailand, 8 Malaria Research Programme, College of Public Health Science, Chulalongkorn University, Bangkok, OPEN ACCESS Thailand, 9 Department of Biology, Faculty of Science, Chulalongkorn University, Bangkok, Thailand, Citation: Tonkin-Hill -

Knowledgecity
#KNOWLEDGECITY MELBOURNE.VIC.GOV.AU/KNOWLEDGEWEEK MELBOURNE KNOWLEDGE WEEK 2014 WELCOME Welcome to the City of Melbourne’s fifth annual Melbourne Knowledge Week. Melbourne Knowledge Week celebrates our multi-billion dollar knowledge sector and the ideas and talent that have Robert Doyle earned Melbourne the title of world’s Lord Mayor most admired knowledge city for a third time. This year’s Melbourne Knowledge Week program will feature more than 90 events including exhibitions, tours, panel discussions, challenges and workshops. It will showcase our expertise in a range of fields including education and research, biotechnology, Cr Jackie Watts advanced manufacturing, information Chair, Knowledge City and communication technology and our creative sector. We encourage you to participate in Melbourne Knowledge Week and learn more about the exciting and innovative work that is shaping our great city. 2 OVERVIEW Melbourne Knowledge Week (MKW) supports and celebrates the innovation, creativity, idea-sharing and thought-leadership that contributes to Melbourne being one of the leading knowledge cities in the world. We have partnered with a range And for the first time we have our own of organisations, businesses and Knowledge Hub on Swanston Street individuals to create a showcase where you can take part in a variety of exclusive opportunities and of extra events or find out what’s experiences which will expand horizons happening throughout the week. and broaden minds across the city. This is a great opportunity for Lots of events are free and there is everyone, to discover, celebrate and something for everyone. contribute to the wealth of knowledge in our community. -

2020 Annual Report PHOTOGRAPH CREDITS: Pp
Response and Recovery 2020 Annual Report PHOTOGRAPH CREDITS: pp. 13/18/back cover, Brett Summerell; p. 19, CDC/Alissa Eckert, MSMI; Dan Higgins, MAMS. Contents Chairman’s 2019/2020 Bioplatforms report Highlights Australia 2 4 5 Bioplatforms Bioplatforms Feature network activities stories 6 7 8 Network National Collaboration LEADERSHIP access Initiatives and partnerships Bioplatforms Australia is committed to maintaining a high standard of governance and leadership. Strategic direction and operational oversight is provided by an independent Board of Directors and supported by an Executive Management Committee who advise on platform technologies and organisational initiatives. Board Members and Responsibilities Executive Management Committee 10 13 16 Bioplatforms Australia’s Directors offer a wealth of experience The Executive Management Committee manages and advises across scientific, business and government domains. on platform issues and operations. It is also responsible for Industry Capabilities BioCommons Each Director has responsibility for particular aspects of implementing strategic initiatives, including Commonwealth engagement network organisational strategy in addition to their fiduciary duties. funding agreements established with network partners. Dr Leslie Trudzik – Chairman Committee members are: Les is a founding Board Member of Bioplatforms Australia and Chair became Chairman in 2013. He is responsible for developing the Andrew Gilbert, Chief Executive, Bioplatforms Australia organisation’s performance and impact framework. -

Melbourne Knowledge Precincts
1 CITY/BIOMEDICAL PRECINCT > Aikenhead Centre for Medical Discovery MELBOURNE > Bio21 Institute > Bionics Institute > Centre for Eye Research Australia (CERA) KNOWLEDGE > Commonwealth Serum Laboratories (CSL) > CSIRO Materials Science and Engineering Parkville > IBM Research Collaboratory for Life Sciences PRECINCTS > Institute for a Broadband Enabled Society (IBES) > Ludwig Institute for Cancer Research > Melbourne Brain Centre > Mental Health Research Institute > National Ageing Research Institute (NARI) > National ICT Australia: Victorian Research Laboratory > O’Brien Institute of Microsurgery Melbourne is home to three knowledge > Orygen Youth Health Centre precincts bringing together academic > Peter Doherty Institute for Infection and Immunity institutions, research and medical > Peter MacCallum Cancer Centre > Royal Melbourne Institute of Technology (RMIT) centres and industry to foster world- > St Vincent’s Institute of Medical Research and leading innovation. St Vincent’s Health > Super Computer Initiative > The Royal Children’s Hospital > The Royal Melbourne Hospital > The Royal Women’s Hospital > University of Melbourne NORTHERN PRECINCT > Victorian Comprehensive Cancer Centre (VCCC) - Opens in 2015 > Victorian Life Sciences Computation Initiative (VLSCI) 3 > Walter and Eliza Hall Institute of Medical Research (WEHI) 2 SOUTH EAST MELBOURNE INNOVATION PRECINCT (SEMIP) CITY/BIOMEDICAL PRECINCT > Australian Synchrotron > Commonwealth Scientific and Industrial Research 1 Organisation (CSIRO) > Melbourne Centre for Nanofabrication