Understadning the Regulation of Endogenous Trpv2 by Growth Factors in Neuronal Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ankrd9 Is a Metabolically-Controlled Regulator of Impdh2 Abundance and Macro-Assembly
ANKRD9 IS A METABOLICALLY-CONTROLLED REGULATOR OF IMPDH2 ABUNDANCE AND MACRO-ASSEMBLY by Dawn Hayward A dissertation submitted to The Johns Hopkins University in conformity with the requirements of the degree of Doctor of Philosophy Baltimore, Maryland April 2019 ABSTRACT Members of a large family of Ankyrin Repeat Domains proteins (ANKRD) regulate numerous cellular processes by binding and changing properties of specific protein targets. We show that interactions with a target protein and the functional outcomes can be markedly altered by cells’ metabolic state. ANKRD9 facilitates degradation of inosine monophosphate dehydrogenase 2 (IMPDH2), the rate-limiting enzyme in GTP biosynthesis. Under basal conditions ANKRD9 is largely segregated from the cytosolic IMPDH2 by binding to vesicles. Upon nutrient limitation, ANKRD9 loses association with vesicles and assembles with IMPDH2 into rod-like structures, in which IMPDH2 is stable. Inhibition of IMPDH2 with Ribavirin favors ANKRD9 binding to rods. The IMPDH2/ANKRD9 assembly is reversed by guanosine, which restores association of ANKRD9 with vesicles. The conserved Cys109Cys110 motif in ANKRD9 is required for the vesicles-to-rods transition as well as binding and regulation of IMPDH2. ANKRD9 knockdown increases IMPDH2 levels and prevents formation of IMPDH2 rods upon nutrient limitation. Thus, the status of guanosine pools affects the mode of ANKRD9 action towards IMPDH2. Advisor: Dr. Svetlana Lutsenko, Department of Physiology, Johns Hopkins University School of Medicine Second reader: -

Repeat Proteins Challenge the Concept of Structural Domains
844 Biochemical Society Transactions (2015) Volume 43, part 5 Repeat proteins challenge the concept of structural domains Rocıo´ Espada*1, R. Gonzalo Parra*1, Manfred J. Sippl†, Thierry Mora‡, Aleksandra M. Walczak§ and Diego U. Ferreiro*2 *Protein Physiology Lab, Dep de Qu´ımica Biologica, ´ Facultad de Ciencias Exactas y Naturales, UBA-CONICET-IQUIBICEN, Buenos Aires, C1430EGA, Argentina †Center of Applied Molecular Engineering, Division of Bioinformatics, Department of Molecular Biology, University of Salzburg, 5020 Salzburg, Austria ‡Laboratoire de physique statistique, CNRS, UPMC and Ecole normale superieure, ´ 24 rue Lhomond, 75005 Paris, France §Laboratoire de physique theorique, ´ CNRS, UPMC and Ecole normale superieure, ´ 24 rue Lhomond, 75005 Paris, France Abstract Structural domains are believed to be modules within proteins that can fold and function independently. Some proteins show tandem repetitions of apparent modular structure that do not fold independently, but rather co-operate in stabilizing structural forms that comprise several repeat-units. For many natural repeat-proteins, it has been shown that weak energetic links between repeats lead to the breakdown of co-operativity and the appearance of folding sub-domains within an apparently regular repeat array. The quasi-1D architecture of repeat-proteins is crucial in detailing how the local energetic balances can modulate the folding dynamics of these proteins, which can be related to the physiological behaviour of these ubiquitous biological systems. Introduction and between repeats, challenging the concept of structural It was early on noted that many natural proteins typically domain. collapse stretches of amino acid chains into compact units, defining structural domains [1]. These domains typically correlate with biological activities and many modern proteins can be described as composed by novel ‘domain arrange- Definition of the repeat-units ments’ [2]. -

A Structural Guide to Proteins of the NF-Kb Signaling Module
Downloaded from http://cshperspectives.cshlp.org/ on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press A Structural Guide to Proteins of the NF-kB Signaling Module Tom Huxford1 and Gourisankar Ghosh2 1Department of Chemistry and Biochemistry, San Diego State University, 5500 Campanile Drive, San Diego, California 92182-1030 2Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093-0375 Correspondence: [email protected] The prosurvival transcription factor NF-kB specifically binds promoter DNA to activate target gene expression. NF-kB is regulated through interactions with IkB inhibitor proteins. Active proteolysis of these IkB proteins is, in turn, under the control of the IkB kinase complex (IKK). Together, these three molecules form the NF-kB signaling module. Studies aimed at charac- terizing the molecular mechanisms of NF-kB, IkB, and IKK in terms of their three-dimen- sional structures have lead to a greater understanding of this vital transcription factor system. F-kB is a master transcription factor that from the perspective of their three-dimensional Nresponds to diverse cell stimuli by activat- structures. ing the expression of stress response genes. Multiple signals, including cytokines, growth factors, engagement of the T-cell receptor, and NF-kB bacterial and viral products, induce NF-kB Introduction to NF-kB transcriptional activity (Hayden and Ghosh 2008). A point of convergence for the myriad NF-kB was discovered in the laboratory of of NF-kB inducing signals is the IkB kinase David Baltimore as a nuclear activity with bind- complex (IKK). Active IKK in turn controls ing specificity toward a ten-base-pair DNA transcription factor NF-kB by regulating pro- sequence 50-GGGACTTTCC-30 present within teolysis of the IkB inhibitor protein (Fig. -
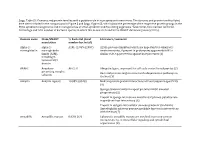
Supp. Table S2: Domains and Protein Families with a Putative Role in Host-Symbiont Interactions
Supp. Table S2: Domains and protein families with a putative role in host-symbiont interactions. The domains and protein families listed here were included in the comparisons in Figure 5 and Supp. Figure S5, which show the percentage of the respective protein groups in the Riftia symbiont metagenome and in metagenomes of other symbiotic and free-living organisms. % bacterial, total number bacterial: Percentage and total number of bacterial species in which this domain is found in the SMART database (January 2019). Domain name Pfam/SMART % bacterial (total Literature/comment annotation number bacterial) Alpha-2- alpha-2- A2M: 42.05% (2057) A2Ms: protease inhibitors which are important for eukaryotic macroglobulin macroglobulin innate immunity, if present in prokaryotes apparently fulfill a family (A2M), similar role, e.g. protection against host proteases (1) including N- terminal MG1 domain ANAPC Anaphase- APC2: 0 Ubiquitin ligase, important for cell cycle control in eukaryotes (2) promoting complex Bacterial proteins might interact with ubiquitination pathways in subunits the host (3) Ankyrin Ankyrin repeats 10.88% (8348) Mediate protein-protein interactions without sequence specificity (4) Sponge symbiont ankyrin-repeat proteins inhibit amoebal phagocytosis (5) Present in sponge microbiome metatranscriptomes, putative role in symbiont-host interactions (6) Present in obligate intracellular amoeba symbiont Candidatus Amoebophilus asiaticus genome, probable function in interactions with the host (7) Armadillo Armadillo repeats 0.83% (67) -

Cryo-EM Structure of the Polycystic Kidney Disease-Like Channel PKD2L1
ARTICLE DOI: 10.1038/s41467-018-03606-0 OPEN Cryo-EM structure of the polycystic kidney disease-like channel PKD2L1 Qiang Su1,2,3, Feizhuo Hu1,3,4, Yuxia Liu4,5,6,7, Xiaofei Ge1,2, Changlin Mei8, Shengqiang Yu8, Aiwen Shen8, Qiang Zhou1,3,4,9, Chuangye Yan1,2,3,9, Jianlin Lei 1,2,3, Yanqing Zhang1,2,3,9, Xiaodong Liu2,4,5,6,7 & Tingliang Wang1,3,4,9 PKD2L1, also termed TRPP3 from the TRPP subfamily (polycystic TRP channels), is involved 1234567890():,; in the sour sensation and other pH-dependent processes. PKD2L1 is believed to be a non- selective cation channel that can be regulated by voltage, protons, and calcium. Despite its considerable importance, the molecular mechanisms underlying PKD2L1 regulations are largely unknown. Here, we determine the PKD2L1 atomic structure at 3.38 Å resolution by cryo-electron microscopy, whereby side chains of nearly all residues are assigned. Unlike its ortholog PKD2, the pore helix (PH) and transmembrane segment 6 (S6) of PKD2L1, which are involved in upper and lower-gate opening, adopt an open conformation. Structural comparisons of PKD2L1 with a PKD2-based homologous model indicate that the pore domain dilation is coupled to conformational changes of voltage-sensing domains (VSDs) via a series of π–π interactions, suggesting a potential PKD2L1 gating mechanism. 1 Ministry of Education Key Laboratory of Protein Science, Tsinghua University, Beijing 100084, China. 2 School of Life Sciences, Tsinghua University, Beijing 100084, China. 3 Beijing Advanced Innovation Center for Structural Biology, Tsinghua University, Beijing 100084, China. 4 School of Medicine, Tsinghua University, Beijing 100084, China. -

Macromolecular Assembly of Polycystin-2 Intracytosolic C-Terminal Domain
Macromolecular assembly of polycystin-2 intracytosolic C-terminal domain Frederico M. Ferreiraa,b,1, Leandro C. Oliveirac, Gregory G. Germinod, José N. Onuchicc,1, and Luiz F. Onuchica,1 aDivision of Nephrology, University of São Paulo School of Medicine, 01246-903, São Paulo, Brazil; bLaboratory of Immunology, Heart Institute, University of São Paulo School of Medicine, 05403-900, São Paulo, Brazil; cCenter for Theoretical Biological Physics, University of California at San Diego, La Jolla, CA 92093; dNational Institute of Diabetes, Digestive, and Kidney Diseases, Bethesda, MD 20892-2560 Contributed by José N. Onuchic, April 28, 2011 (sent for review March 20, 2011) Mutations in PKD2 are responsible for approximately 15% of the In spite of the aforementioned information and insights, the autosomal dominant polycystic kidney disease cases. This gene macromolecular assembly of PC2t homooligomer continued to encodes polycystin-2, a calcium-permeable cation channel whose be an open question. In the current work, we present the most C-terminal intracytosolic tail (PC2t) plays an important role in its comprehensive set of analyses yet performed and that show interaction with a number of different proteins. In the present PC2t forms a homotetrameric oligomer. We have proposed a study, we have comprehensively evaluated the macromolecular PC2 C-terminal domain delimitation and submitted it to a range assembly of PC2t homooligomer using a series of biophysical of biochemical and biophysical evaluations, including chemical and biochemical analyses. Our studies, based on a new delimitation cross-linking, dynamic light scattering (DLS), circular dichroism of PC2t, have revealed that it is capable of assembling as a homo- (CD) and small angle X-ray scattering (SAXS) analyses. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

Data-Driven Analysis of TRP Channels in Cancer
CANCER GENOMICS & PROTEOMICS 13 : 83-90 (2016) Data-driven Analysis of TRP Channels in Cancer: Linking Variation in Gene Expression to Clinical Significance YU RANG PARK 1* , JUNG NYEO CHUN 2* , INSUK SO 2, HWA JUNG KIM 3, SEUNGHEE BAEK 4, JU-HONG JEON 2 and SOO-YONG SHIN 1,5 1Office of Clinical Research Information, and Departments of 3Clinical Epidemiology and Biostatistics, and 5Biomedical Informatics, Asan Medical Center, Seoul, Republic of Korea; 2Department of Physiology and Biomedical Sciences, Institute of Human-Environment Interface Biology, Seoul National University College of Medicine, Seoul, Republic of Korea; 4Department of Preventive Medicine, University of Ulsan College of Medicine, Seoul, Republic of Korea Abstract. Background: Experimental evidence has intracellular Ca 2+ in response to various internal and external suggested that transient receptor potential (TRP) channels stimuli (1, 2). In human, the TRP channel superfamily play a crucial role in tumor biology. However, clinical consists of 27 isotypes that are classified into six subfamilies relevance and significance of TRP channels in cancer remain (3): canonical (TRPC), vanilloid (TRPV), melastatin (TRPM), largely unknown. Materials and Methods: We applied a data- polycystin (TRPP), mucolipin (TRPML), and ankyrin driven approach to dissect the expression landscape of 27 (TRPA). Emerging evidence has shown that the aberrant TRP channel genes in 14 types of human cancer using functions of TRP channels are closely associated with cancer International Cancer Genome Consortium data. Results: hallmarks, such as sustaining proliferative signaling, evading TRPM2 was found overexpressed in most tumors, whereas growth suppressors, resisting cell death, and activating TRPM3 was broadly down-regulated. TRPV4 and TRPA1 invasion and metastasis (4, 5). -

WSB1: from Homeostasis to Hypoxia Moinul Haque1,2,3, Joseph Keith Kendal1,2,3, Ryan Matthew Macisaac1,2,3 and Douglas James Demetrick1,2,3,4*
Haque et al. Journal of Biomedical Science (2016) 23:61 DOI 10.1186/s12929-016-0270-3 REVIEW Open Access WSB1: from homeostasis to hypoxia Moinul Haque1,2,3, Joseph Keith Kendal1,2,3, Ryan Matthew MacIsaac1,2,3 and Douglas James Demetrick1,2,3,4* Abstract The wsb1 gene has been identified to be important in developmental biology and cancer. A complex transcriptional regulation of wsb1 yields at least three functional transcripts. The major expressed isoform, WSB1 protein, is a substrate recognition protein within an E3 ubiquitin ligase, with the capability to bind diverse targets and mediate ubiquitinylation and proteolytic degradation. Recent data suggests a new role for WSB1 as a component of a neuroprotective pathway which results in modification and aggregation of neurotoxic proteins such as LRRK2 in Parkinson’s Disease, via an unusual mode of protein ubiquitinylation. WSB1 is also involved in thyroid hormone homeostasis, immune regulation and cellular metabolism, particularly glucose metabolism and hypoxia. In hypoxia, wsb1 is a HIF-1 target, and is a regulator of the degradation of diverse proteins associated with the cellular response to hypoxia, including HIPK2, RhoGDI2 and VHL. Major roles are to both protect HIF-1 function through degradation of VHL, and decrease apoptosis through degradation of HIPK2. These activities suggest a role for wsb1 in cancer cell proliferation and metastasis. As well, recent work has identified a role for WSB1 in glucose metabolism, and perhaps in mediating the Warburg effect in cancer cells by maintaining the function of HIF1. Furthermore, studies of cancer specimens have identified dysregulation of wsb1 associated with several types of cancer, suggesting a biologically relevant role in cancer development and/or progression. -

The Heteromeric PC-1/PC-2 Polycystin Complex Is Activated by the PC-1 N-Terminus
RESEARCH ARTICLE The heteromeric PC-1/PC-2 polycystin complex is activated by the PC-1 N-terminus Kotdaji Ha1, Mai Nobuhara1, Qinzhe Wang2, Rebecca V Walker3, Feng Qian3, Christoph Schartner1, Erhu Cao2, Markus Delling1* 1Department of Physiology, University of California, San Francisco, San Francisco, United States; 2Department of Biochemistry, University of Utah School of Medicine, Salt Lake City, United States; 3Division of Nephrology, Department of Medicine, University of Maryland School of Medicine, Baltimore, United States Abstract Mutations in the polycystin proteins, PC-1 and PC-2, result in autosomal dominant polycystic kidney disease (ADPKD) and ultimately renal failure. PC-1 and PC-2 enrich on primary cilia, where they are thought to form a heteromeric ion channel complex. However, a functional understanding of the putative PC-1/PC-2 polycystin complex is lacking due to technical hurdles in reliably measuring its activity. Here we successfully reconstitute the PC-1/PC-2 complex in the plasma membrane of mammalian cells and show that it functions as an outwardly rectifying channel. Using both reconstituted and ciliary polycystin channels, we further show that a soluble fragment generated from the N-terminal extracellular domain of PC-1 functions as an intrinsic agonist that is necessary and sufficient for channel activation. We thus propose that autoproteolytic cleavage of the N-terminus of PC-1, a hotspot for ADPKD mutations, produces a soluble ligand in vivo. These findings establish a mechanistic framework for understanding the role of PC-1/PC-2 heteromers in ADPKD and suggest new therapeutic strategies that would expand upon the limited symptomatic treatments currently available for this progressive, terminal disease. -

Opening TRPP2 (PKD2L1) Requires the Transfer of Gating Charges
Opening TRPP2 (PKD2L1) requires the transfer of gating charges Leo C. T. Nga, Thuy N. Viena, Vladimir Yarov-Yarovoyb, and Paul G. DeCaena,1 aDepartment of Pharmacology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611; and bDepartment of Physiology and Membrane Biology, University of California, Davis, CA 95616 Edited by Richard W. Aldrich, The University of Texas at Austin, Austin, TX, and approved June 19, 2019 (received for review February 18, 2019) The opening of voltage-gated ion channels is initiated by transfer their ligands (exogenous or endogenous) is sufficient to initiate of gating charges that sense the electric field across the mem- channel opening. Although TRP channels share a similar to- brane. Although transient receptor potential ion channels (TRP) pology with most VGICs, few are intrinsically voltage gated, and are members of this family, their opening is not intrinsically linked most do not have gating charges within their VSD-like domains to membrane potential, and they are generally not considered (16). Current conducted by TRP family members is often rectifying, voltage gated. Here we demonstrate that TRPP2, a member of the but this form of voltage dependence is usually attributed to divalent polycystin subfamily of TRP channels encoded by the PKD2L1 block or other conditional effects (17, 18). There are 3 members of gene, is an exception to this rule. TRPP2 borrows a biophysical riff the polycystin subclass: TRPP1 (PKD2 or polycystin-2), TRPP2 from canonical voltage-gated ion channels, using 2 gating charges (PKD2-L1 or polycystin-L), and TRPP3 (PKD2-L2). TRPP1 is the found in its fourth transmembrane segment (S4) to control its con- founding member of this family, and variants in the PKD2 gene ductive state. -

Drug Discovery for Polycystic Kidney Disease
Acta Pharmacologica Sinica (2011) 32: 805–816 npg © 2011 CPS and SIMM All rights reserved 1671-4083/11 $32.00 www.nature.com/aps Review Drug discovery for polycystic kidney disease Ying SUN, Hong ZHOU, Bao-xue YANG* Department of Pharmacology, School of Basic Medical Sciences, Peking University, and Key Laboratory of Molecular Cardiovascular Sciences, Ministry of Education, Beijing 100191, China In polycystic kidney disease (PKD), a most common human genetic diseases, fluid-filled cysts displace normal renal tubules and cause end-stage renal failure. PKD is a serious and costly disorder. There is no available therapy that prevents or slows down the cystogen- esis and cyst expansion in PKD. Numerous efforts have been made to find drug targets and the candidate drugs to treat PKD. Recent studies have defined the mechanisms underlying PKD and new therapies directed toward them. In this review article, we summarize the pathogenesis of PKD, possible drug targets, available PKD models for screening and evaluating new drugs as well as candidate drugs that are being developed. Keywords: polycystic kidney disease; drug discovery; kidney; candidate drugs; animal model Acta Pharmacologica Sinica (2011) 32: 805–816; doi: 10.1038/aps.2011.29 Introduction the segments of the nephron. Autosomal recessive polycystic Polycystic kidney disease (PKD), an inherited human renal kidney disease (ARPKD) results primarily from the mutations disease, is characterized by massive enlargement of fluid- in a single gene, Pkhd1[14]. Its frequency is estimated to be filled renal tubular and/or collecting duct cysts[1]. Progres- one per 20000 individuals. The PKHD1 protein, fibrocystin, sively enlarging cysts compromise normal renal parenchyma, has been found to be localized to primary cilia and the basal often leading to renal failure.