Verdazyl Radicals As Substrates for Organic Synthesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Dictionary of Chinese Characters: Accessed by Phonetics
A dictionary of Chinese characters ‘The whole thrust of the work is that it is more helpful to learners of Chinese characters to see them in terms of sound, than in visual terms. It is a radical, provocative and constructive idea.’ Dr Valerie Pellatt, University of Newcastle. By arranging frequently used characters under the phonetic element they have in common, rather than only under their radical, the Dictionary encourages the student to link characters according to their phonetic. The system of cross refer- encing then allows the student to find easily all the characters in the Dictionary which have the same phonetic element, thus helping to fix in the memory the link between a character and its sound and meaning. More controversially, the book aims to alleviate the confusion that similar looking characters can cause by printing them alongside each other. All characters are given in both their traditional and simplified forms. Appendix A clarifies the choice of characters listed while Appendix B provides a list of the radicals with detailed comments on usage. The Dictionary has a full pinyin and radical index. This innovative resource will be an excellent study-aid for students with a basic grasp of Chinese, whether they are studying with a teacher or learning on their own. Dr Stewart Paton was Head of the Department of Languages at Heriot-Watt University, Edinburgh, from 1976 to 1981. A dictionary of Chinese characters Accessed by phonetics Stewart Paton First published 2008 by Routledge 2 Park Square, Milton Park, Abingdon, OX14 4RN Simultaneously published in the USA and Canada by Routledge 270 Madison Ave, New York, NY 10016 Routledge is an imprint of the Taylor & Francis Group, an informa business This edition published in the Taylor & Francis e-Library, 2008. -

"Radical Stability --- Thermochemical Aspects" In
Radical Stability—Thermochemical Aspects Johnny Hioe and Hendrik Zipse Department of Chemistry, LMU M¨unchen, M¨unchen, Germany 1 INTRODUCTION is quite challenging. Kinetic data, in contrast, are much more difficult to predict by theory, while the The terms “transient” and “persistent” are used determination of reaction rates can be approached frequently in the scientific literature to describe experimentally with a variety of direct or indirect the kinetic properties of open shell systems in methods, at least for sufficiently fast reactions homogeneous solution.1–5 The hydroxyl radical (see Radical Kinetics and Clocks). Theory and (HO•, 1), for example, is a transient species of experiment pair up nicely in this respect, as a central importance in atmospheric chemistry (see combination of these approaches is able to provide Atmospheric Radical Chemistry), as well as one a comprehensive picture of thermodynamic and of the most important reactive oxygen species kinetic data. (ROS) in aqueous solution, whereas the nitroxide 2,2,6,6-tetramethylpiperidine-1-oxyl, TEMPO (2)is a persistent radical stable enough to be bottled and 2 DEFINITIONS OF RADICAL STABILITY sold in bulk (Figure 1) (see Nitroxides in Synthetic Radical Chemistry). The thermodynamic stability of C-centered radicals However, despite their widespread use, these can be defined in various ways and several options terms are not too helpful for a quantitative approach are discussed in the following.6–10 One of the to radical chemistry as they do not reflect the most often used definitions is based on hydrogen influence of thermochemical driving force and transfer reactions as shown in Scheme 1 for reaction • intrinsic reaction barrier on the observed lifetime. -

Functionalised Oximes: Emergent Precursors for Carbon-, Nitrogen- and Oxygen-Centred Radicals
molecules Review Functionalised Oximes: Emergent Precursors for Carbon-, Nitrogen- and Oxygen-Centred Radicals John C. Walton Received: 11 December 2015 ; Accepted: 30 December 2015 ; Published: 7 January 2016 Academic Editor: Roman Dembinski EaStCHEM School of Chemistry, University of St. Andrews, St. Andrews, Fife KY16 9ST, UK; [email protected]; Tel.: +44-01334-463-864; Fax: +44-01334-463-808 Abstract: Oxime derivatives are easily made, are non-hazardous and have long shelf lives. They contain weak N–O bonds that undergo homolytic scission, on appropriate thermal or photochemical stimulus, to initially release a pair of N- and O-centred radicals. This article reviews the use of these precursors for studying the structures, reactions and kinetics of the released radicals. Two classes have been exploited for radical generation; one comprises carbonyl oximes, principally oxime esters and amides, and the second comprises oxime ethers. Both classes release an iminyl radical together with an equal amount of a second oxygen-centred radical. The O-centred radicals derived from carbonyl oximes decarboxylate giving access to a variety of carbon-centred and nitrogen-centred species. Methods developed for homolytically dissociating the oxime derivatives include UV irradiation, conventional thermal and microwave heating. Photoredox catalytic methods succeed well with specially functionalised oximes and this aspect is also reviewed. Attention is also drawn to the key contributions made by EPR spectroscopy, aided by DFT computations, in elucidating the structures and dynamics of the transient intermediates. Keywords: oxime esters; oxime ethers; free radicals; organic synthesis; photochemical reactions; EPR spectroscopy; photoredox catalysis; N-heterocycles 1. Introduction A huge variety of compounds containing the carbonyl functional group is available from natural and commercial sources. -
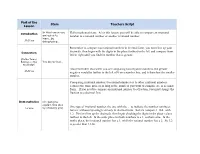
Part of the Lesson Stem Teachers Script Hello Mathematicians. After
Part of the Stem Teachers Script Lesson In this lesson you Introduction Hello mathematicians. After this lesson, you will be able to compare an irrational are going to number to a rational number or another irrational number. learn…by 10-20 sec doing/using… Remember to compare two rational numbers in decimal form, you must line up your decimals, then begin with the digits in the place furthest to the left and compare from Connection left to right until you find the number that is greater. (Define Terms/ Building on Prior You know that… Knowledge) Also remember, that when you are comparing two negative numbers, the greater 30-60 sec negative would be further to the left of 0 on a number line, and is therefore the smaller number. Comparing irrational numbers to rational numbers or to other irrational numbers requires the same process as long as the numbers you want to compare are in decimal form. If you need to compare an irrational number to a fraction, you must change the fraction to a decimal first. Demonstration I’m going to explain this idea One type of irrational number, the one with the ... to indicate the number continues 1-3 minc by showing you¦ forever without repeating is already in decimal form. So let's compare 1.124...with 1.2. First we line up the decimals, then begin checking the digits in the place values furthest to the left. In the units place for both numbers is a 1, so that's a tie. In the tenths place, the irrational number has a 1, while the rational number has a 2. -

Formation and Structure of Radicals from D-Ribose and 2-Deoxy- D-Ribose by Reactions with SO 4 Radicals in Aqueous Solution
Formation and Structure of Radicals from D-Ribose and 2-Deoxy- D-ribose by Reactions with SO 4 Radicals in Aqueous Solution. An in-situ Electron Spin Resonance Study Janko N. Herak Faculty of Pharmacy and Biochemistry, The University of Zagreb, Zagreb. Croatia, Yugoslavia Günter Behrens Max-Planck-Institut für Strahlenchemie. D-4330 Mülheim a.d. Ruhr, Bundesrepublik Deutschland Z. Naturforsch. 41c, 1062 — 1068 (1986); received January 31, 1986 Free Radicals, Deoxy-D-ribose, D-Ribose, Conformational Kinetics, Electron Spin Resonance ESR spectroscopy has been used to analyse the conformation of the radicals produced by the reaction of SOj with D-ribose (1),and 2-deoxy-D-ribose (6 ),at pH 1.3-5. From ribose three different types of radicals formed by H abstraction at C-l, C-2 and C-3 followed by a regio- selective a,ß-water elimination have been identified: the 2-deoxy-ribonolacton-2-yl (3), the 1-deoxy-pentopyranos-2-ulos-l-yl (4). and the 4-deoxy-pentopyranos-3-ulos-4-yl (2). Using deoxyribose two radicals of similar type, formed by H abstraction at C-3 and C-4 followed by water elimination, have been observed: the 2,4-dideoxy-3-ulos-4-yl (7) and the 2,3-dideoxy-4- ulos-3-yl (8). In addition, from both sugars an a-hydroxyalkyl radical has been identified based in part on the timing of their conformational motions: the ribos-3-yl (5) (the precursor of 2) and the 2-deoxy-ribos-l-yl (9), respectively. For radical 5 the rate constant k(e) for the water elimination and hence transformation into radical 2 was estimated. -
Towards New Approaches for the Generation of Phosphorus Based Radical: Synthetic and Mechanistic Investigations
Towards new approaches for the generation of phosphorus based radical : synthetic and mechanistic investigations Giovanni Fausti To cite this version: Giovanni Fausti. Towards new approaches for the generation of phosphorus based radical : synthetic and mechanistic investigations. Organic chemistry. Normandie Université, 2017. English. NNT : 2017NORMC271. tel-01893162 HAL Id: tel-01893162 https://tel.archives-ouvertes.fr/tel-01893162 Submitted on 11 Oct 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour obtenir le diplôme de doctorat Spécialité CHIMIE Préparé au sein de l'Université de Caen Normandie Τοwards new apprοaches fοr the generatiοn οf phοsphοrus based radical : synthetic and mechanistic investigatiοns Présentée et soutenue par Giovanni FAUSTI Thèse soutenue publiquement le 24/07/2017 devant le jury composé de Professeur des universités, UNIVERSITE PARIS 5 UNIVERSITE M. PHILIPPE BELMONT Rapporteur du jury PARIS DESC Maître de conférences HDR, UNIVERSITE TOULOUSE 3 PAUL Mme GHENWA BOUHADIR Rapporteur du jury SABATIER Mme ISABELLE GILLAIZEAU Professeur des universités, UNIVERSITE ORLEANS Président du jury M. SAMI LADHDAR Chargé de recherche, UNIVERSITE CAEN NORMANDIE Membre du jury Mme ANNIE-CLAUDE GAUMONT Professeur des universités, UNIVERSITE CAEN NORMANDIE Directeur de thèse Thèse dirigée par ANNIE-CLAUDE GAUMONT, Laboratoire de chimie moléculaire et thio-organique (Caen) TABLE OF CONTENTS List of tables………………………………………………………………………. -
Radical Addition to Isonitriles: a Route to Polyfunctionalized Alkenes Through a Novel Three-Component Radical Cascade Reaction
J. Org. Chem. 2000, 65, 2763-2772 2763 Radical Addition to Isonitriles: A Route to Polyfunctionalized Alkenes through a Novel Three-Component Radical Cascade Reaction Rino Leardini, Daniele Nanni,* and Giuseppe Zanardi Dipartimento di Chimica Organica “A. Mangini”, Universita` di Bologna, Viale Risorgimento 4, I-40136 Bologna, Italy Received December 6, 1999 The reaction of aromatic disulfides, alkynes, and isonitriles under photolytic conditions affords polyfunctionalized alkenessâ-arylthio-substituted acrylamides or acrylonitrilessin fair yields through a novel three-component radical cascade reaction. The procedure entails addition of a sulfanyl radical to the alkyne followed by attack of the resulting vinyl radical to the isonitrile. A fast reaction, e.g., scavenging by a nitro derivative or â-fragmentation, is necessary in order to trap the final imidoyl radical, since addition of vinyl radicals to isonitriles seems to be a reversible process. The stereochemistry of the reaction is discussed, particularly with respect to the stereochemical outcome of related hydrogen abstraction reactions by the same vinyl radicals. The lower or even inverted preference for either geometrical isomer observed in our cases with respect to that encountered in hydrogen abstraction reactions is explained in terms of transition-state interactions and/or isomerization of the final imidoyl radical. The latter possibility is supported by semiempirical calculations, which show that the spin distribution in the imidoyl radical can allow rotation of the adjacent -

The Regiochemistry of Alkenylsilyl, Alkenyldisilanyl and Alkenylsilyloxy Radical Cyclîzations
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1984 The egr iochemistry of alkenylsilyl, alkenyldisilanyl and alkenylsilyloxy radical cyclizations Anthony Revis Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Revis, Anthony, "The er giochemistry of alkenylsilyl, alkenyldisilanyl and alkenylsilyloxy radical cyclizations " (1984). Retrospective Theses and Dissertations. 9023. https://lib.dr.iastate.edu/rtd/9023 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This reproduction was made from a copy of a document sent to us for microfilming. While the most advanced technology has been used to photograph and reproduce this document, the quahty of the reproduction is heavily dependent upon the quality of the material submitted. The following explanation of techniques is provided to help clarify markings or notations which may appear on this reproduction. 1.The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting through an image and duplicating adjacent pages to assure complete continuity. 2. When an image on the film is obhterated with a round black mark, it is an indication of either blurred copy because of movement during exposure, duplicate copy, or copyrighted materials that should not have been filmed. -
Tocopheroxyl Radical 1
77:222 Spring 2003 Free Radicals in Biology and Medicine Page 0 This student paper was written as an assignment in the graduate course Free Radicals in Biology and Medicine (77:222, Spring 2003) offered by the Free Radical and Radiation Biology Program B-180 Med Labs The University of Iowa Iowa City, IA 52242-1181 Spring 2003 Term Instructors: GARRY R. BUETTNER, Ph.D. LARRY W. OBERLEY, Ph.D. with guest lectures from: Drs. Freya Q . Schafer, Douglas R. Spitz, and Frederick E. Domann The Fine Print: Because this is a paper written by a beginning student as an assignment, there are no guarantees that everything is absolutely correct and accurate. In view of the possibility of human error or changes in our knowledge due to continued research, neither the author nor The University of Iowa nor any other party who has been involved in the preparation or publication of this work warrants that the information contained herein is in every respect accurate or complete, and they are not responsible for any errors or omissions or for the results obtained from the use of such information. Readers are encouraged to confirm the information contained herein with other sources. All material contained in this paper is copyright of the author, or the owner of the source that the material was taken from. This work is not intended as a threat to the ownership of said copyrights. X.M. Zheng Tocopheroxyl Radical 1 Tocopheroxyl Radical by Xiaomei Zheng B-180 Medical Laboratories Department of Free Radical and Radiation Biology The University of Iowa Iowa City, IA 52242-1181 For 77:222, Spring 2003 12, February 2003 Abbreviations: - • AscH , ascorbate; CoQ10H2, the reduced form of coenzyme Q; CoQ10H , coenzyme Q radical; ESR, electron spin resonance; FMN, flavin mononucleotede; EDTA, ethylenediaminetetraacetic acid; GO: glucose oxidase; HRP: horseradish perxidase; LH, bisallylic hydrogen; L•, lipid radical; LOO•, lipid peroxyl radical; LDL: low-density lipoprotein. -

Part of the Lesson Stem Teachers Script Hello Mathematicians. After
Part of the Stem Teachers Script Lesson In this lesson you Introduction Hello mathematicians. After today's lesson, you will be able to simplify expressions are going to involving square roots. learn…by 10-20 sec doing/using… You remember that the square root of a number is the side length of a square with an area equal to that number. So the square root of 16 is 4 because a square with an area of 16 would have a side length of 4. Since all sides of a square are the same Connection length, finding the square root of a number, "A" means finding some number that when multiplied by itself gives you A. So the square root of 25 is 5 because 5 times (Define Terms/ itself is 25. Building on Prior You know that… Knowledge) 30-60 sec Also remember that while all numbers greater than zero have a square root, only numbers called, "perfect squares," have integer square roots. Trying to take the square root of a number that is not a perfect square results in an irrational number. So 18 is not a perfect square because 4 times itself is 16, 5 times itself is 25, which means the square root of 18 is an irrational number greater than 4 but less than 5. Demonstration I’m going to But sometimes it is possible and very useful to simplify square roots into smaller explain this idea terms. We will simplify radical 18 in just a minute, but first I want you to think about 1-3 minc by showing you¦ something. -

Tributyltin Mediated Radical Cyclizations of Enediynes and Their Subsequent Transformation to Fulvenes and Indenes Scott W
Florida State University Libraries Electronic Theses, Treatises and Dissertations The Graduate School 2004 Tributyltin Mediated Radical Cyclizations of Enediynes and Their Subsequent Transformation to Fulvenes and Indenes Scott W. Peabody Follow this and additional works at the FSU Digital Library. For more information, please contact [email protected] THE FLORIDA STATE UNIVERSITY COLLEGE OF ARTS AND SCIENCE TRIBUTYLTIN MEDIATED RADICAL CYCLIZATIONS OF ENEDIYNES AND THEIR SUBSEQUENT TRANSFORMATION TO FULVENES AND INDENES By SCOTT W. PEABODY A Thesis submitted to the Department of Chemistry and Biochemistry in partial fulfillment of the requirements for the degree of Master of Science Degree Awarded: Summer Semester, 2004 The members of the Committee approve the Thesis of Scott W. Peabody defended on April 28, 2002. Dr. Igor Alabugin Professor Directing Thesis Dr. Gregory Dudley Committee Member Dr. Oliver Steinbock Committee Member Approved: Naresh Dalal, Chair, Department of Chemistry and Biochemistry The Office of Graduate Studies has verified and approved the above named committee members. ii This is dedicated to my family and to future wife. iii ACKNOWLEDGEMENTS The author wished to extend his sincere appreciation to Prof. Igor Alabugin, whose patience, guidance and support has been instrumental in all the work that I have conducted. I would further like to thank Dr. Serguei Kovalenko for his day in and day out dedication, supervision and instruction and Dr. Mariappan Manoharan for the computations included in this work. I would also like to thank CDR Richard Sanders, USCG, who gave me my first introduction into chemical research and who has served as a mentor over so many years. -

New Avenues for C–B Bond Formation Via Radical Intermediates Cite This: Chem
Chemical Science PERSPECTIVE View Article Online View Journal | View Issue New avenues for C–B bond formation via radical intermediates Cite this: Chem. Sci.,2019,10,8503 All publication charges for this article Florian W. Friese and Armido Studer * have been paid for by the Royal Society of Chemistry This perspective gives an overview on recent findings in the emerging area of C-radical borylation using diborons as radical trapping reagents. Aryl, vinyl and alkyl boronic esters can be accessed via such an approach under mild conditions. These processes are complementary to established transition metal catalysed cross coupling reactions. Radical borylations can be conducted in the absence of a transition Received 26th July 2019 metal but some processes require transition metals as catalysts. It will be shown that various readily Accepted 3rd September 2019 available C-radical precursors can be used to run these borylations. For a better understanding of the DOI: 10.1039/c9sc03765a chemistry, mechanistic discussions are also presented and an outlook on this topic will be provided at rsc.li/chemical-science the end of the article. Creative Commons Attribution-NonCommercial 3.0 Unported Licence. 1. Introduction Notably, published review articlescoverpartsofthe current research activities in this highly active eld but not in Aryl and alkyl boronates are highly valuable building blocks in its entirety, especially considering the borylation of sp3- chemical synthesis, especially considering their applications centered radicals.8 It is therefore the aim of this perspective to in transition-metal catalysed C–C coupling reactions like the address the whole eld of boronate synthesis using radical Suzuki–Miyaura1 or Chan–Lam reaction.2 Furthermore, the pathways.