The Entner-Doudoroff Pathway and Development of a Promoter-Trapping System
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A New Insight Into Role of Phosphoketolase Pathway in Synechocystis Sp
www.nature.com/scientificreports OPEN A new insight into role of phosphoketolase pathway in Synechocystis sp. PCC 6803 Anushree Bachhar & Jiri Jablonsky* Phosphoketolase (PKET) pathway is predominant in cyanobacteria (around 98%) but current opinion is that it is virtually inactive under autotrophic ambient CO2 condition (AC-auto). This creates an evolutionary paradox due to the existence of PKET pathway in obligatory photoautotrophs. We aim to answer the paradox with the aid of bioinformatic analysis along with metabolic, transcriptomic, fuxomic and mutant data integrated into a multi-level kinetic model. We discussed the problems linked to neglected isozyme, pket2 (sll0529) and inconsistencies towards the explanation of residual fux via PKET pathway in the case of silenced pket1 (slr0453) in Synechocystis sp. PCC 6803. Our in silico analysis showed: (1) 17% fux reduction via RuBisCO for Δpket1 under AC-auto, (2) 11.2–14.3% growth decrease for Δpket2 in turbulent AC-auto, and (3) fux via PKET pathway reaching up to 252% of the fux via phosphoglycerate mutase under AC-auto. All results imply that PKET pathway plays a crucial role under AC-auto by mitigating the decarboxylation occurring in OPP pathway and conversion of pyruvate to acetyl CoA linked to EMP glycolysis under the carbon scarce environment. Finally, our model predicted that PKETs have low afnity to S7P as a substrate. Metabolic engineering of cyanobacteria provides many options for producing valuable compounds, e.g., acetone from Synechococcus elongatus PCC 79421 and butanol from Synechocystis sp. strain PCC 68032. However, certain metabolites or overproduction of intermediates can be lethal. Tere is also a possibility that required mutation(s) might be unstable or the target bacterium may even be able to maintain the fux distribution for optimal growth balance due to redundancies in the metabolic network, such as alternative pathways. -

Sterile Spikelets Assimilate Carbon in Sorghum and Related Grasses
bioRxiv preprint doi: https://doi.org/10.1101/396473; this version posted January 5, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. 1 Sterile spikelets assimilate carbon in sorghum and related grasses Taylor AuBuchon-Elder1,a, Viktoriya Coneva1,a,d, David M. Goad a,b, Doug K. Allen2,a,c,e, and Elizabeth A. Kellogg2,a,e aDonald Danforth Plant Science Center, St. Louis, Missouri, 63132 USA bDepartment of Biology, Washington University, St. Louis, Missouri, 63130 USA cUSDA-ARS, St. Louis, Missouri, 63132 USA 1These authors contributed equally to this work. 2These authors contributed equally to this work. dCurrent address: Kenyon College, Gambier, OH 43022 eAddress correspondence to: [email protected]; [email protected] ORCID IDs: 0000-0002-0640-5135 (V.C.); 0000-0001-8658-6660 (D.M.G.); 0000-0001-8599- 8946 (D.K.A.); 0000-0003-1671-7447 (E.A.K.) Short title: Carbon assimilation in sorghum spikelets The author responsible for distribution of materials integral to the findings presented in this article is Elizabeth A. Kellogg ([email protected]). bioRxiv preprint doi: https://doi.org/10.1101/396473; this version posted January 5, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. -

RESPIRATION Pentose Phosphate Pathway Or Hexose Monophosphate Pathway
RESPIRATION Pentose Phosphate Pathway or Hexose Monophosphate Pathway Pentose phosphate pathway or hexose monophosphate pathway (HMP pathway) is the other common pathway to break down glucose to pyruvate and operates in both aerobic and anaerobic conditions. This pathway produces NADPH, which carries chemical energy in the form of reducing power and is used almost universally as the reductant in anabolic (energy utilization) pathways (e.g., fatty acid biosynthesis, cholesterol biosynthesis, nucleotide biosynthesis) and detoxification pathways (e.g., reduction of oxidized glutathione, cytochrome P450 monooxygenases). Also, the pentose phosphate pathway generates pentose sugar ribose and its derivatives, which are necessary for the biosynthesis of nucleic acids (DNA and RNA) as well as ATP, NADH, FAD, and coenzyme A. In this way, though the pentose phosphate pathway may be a source of energy in many microorganisms, it is more often of greater importance in various biosynthetic pathways. Pentose phosphate pathway consists of two phases: the oxidative phase and the non-oxidative phase. Oxidative Phase: The oxidative phase of the pentose phosphate pathway initiates with the conversion of glucose 6- phosphate to 6-Phosphogluconate. NADP+ is the electron acceptor yielding NADPH during this reaction. 6-Phosphogluconate, a six-carbon sugar, is then oxidativelydecarboxylated to yield ribulose 5-phosphate, a five-carbon sugar. NADP+ is again the electron acceptor yielding NADPH. In the final step of oxidative phase, there is isomerisation of ribulose 5-phosphatc to ribose 5- phosphate by phosphopentose isomerase and the conversion of ribulose 5-phosphate into its epimerxylulose 5-phosphate by phosphopentose epimerase for the transketolase reaction in non- oxidative phase. -
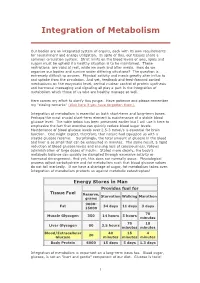
Integration of Metabolism
Integration of Metabolism Our bodies are an integrated system of organs, each with its own requirements for nourishment and energy utilization. In spite of this, our tissues share a common circulation system. Strict limits on the blood levels of ions, lipids and sugars must be upheld if a healthy situation is to be maintained. These restrictions are valid at rest, while we work and after meals. How do we organize our bodies and survive under differing situations? The question is extremely difficult to answer. Physical activity and meals greatly alter influx to and uptake from the circulation. And yet, feedback and feed-forward control mechanisms on the enzymatic level, central nuclear control of protein synthesis and hormonal messaging and signaling all play a part in the integration of metabolism which those of us who are healthy manage so well. Here comes my effort to clarify this jungle. Have patience and please remember my "closing remarks" (click here if you have forgotten them). Integration of metabolism is essential on both short-term and long-term bases. Perhaps the most crucial short-term element is maintenance of a stable blood glucose level. The table below has been presented earlier but I will use it here to emphasize the fact that exercise can quickly reduce blood sugar levels. Maintenance of blood glucose levels over 2.5-3 mmol/s is essential for brain function. One might expect, therefore, that nature had equipped us with a sizable glucose reserve. Surprisingly, the total amount of glucose in the blood and liver is so small that can be exhausted in minutes. -

Diagnostic Value of Serum Enzymes-A Review on Laboratory Investigations
Review Article ISSN 2250-0480 VOL 5/ ISSUE 4/OCT 2015 DIAGNOSTIC VALUE OF SERUM ENZYMES-A REVIEW ON LABORATORY INVESTIGATIONS. 1VIDYA SAGAR, M.SC., 2DR. VANDANA BERRY, MD AND DR.ROHIT J. CHAUDHARY, MD 1Vice Principal, Institute of Allied Health Sciences, Christian Medical College, Ludhiana 2Professor & Ex-Head of Microbiology Christian Medical College, Ludhiana 3Assistant Professor Department of Biochemistry Christian Medical College, Ludhiana ABSTRACT Enzymes are produced intracellularly, and released into the plasma and body fluids, where their activities can be measured by their abilities to accelerate the particular chemical reactions they catalyze. But different serum enzymes are raised when different tissues are damaged. So serum enzyme determination can be used both to detect cellular damage and to suggest its location in situ. Some of the biochemical markers such as alanine aminotransferase, aspartate aminotransferase, alkaline phasphatase, gamma glutamyl transferase, nucleotidase, ceruloplasmin, alpha fetoprotein, amylase, lipase, creatine phosphokinase and lactate dehydrogenase are mentioned to evaluate diseases of liver, pancreas, skeletal muscle, bone, etc. Such enzyme test may assist the physician in diagnosis and treatment. KEYWORDS: Liver Function tests, Serum Amylase, Lipase, CPK and LDH. INTRODUCTION mitochondrial AST is seen in extensive tissue necrosis during myocardial infarction and also in chronic Liver diseases like liver tissue degeneration DIAGNOSTIC SERUM ENZYME and necrosis². But lesser amounts are found in Enzymes are very helpful in the diagnosis of brain, pancreas and lung. Although GPT is plentiful cardiac, hepatic, pancreatic, muscular, skeltal and in the liver and occurs only in the small amount in malignant disorders. Serum for all enzyme tests the other tissues. -

Chapter 23 Gluconeogenesis Gluconeogenesis, Con't
BCH 4054 Spring 2001 Chapter 23 Lecture Notes Slide 1 Chapter 23 Gluconeogenesis Glycogen Metabolism Pentose Phosphate Pathway Slide 2 Gluconeogenesis • Humans use about 160 g of glucose per day, about 75% for the brain. • Body fluids and glycogen stores supply only a little over a day’s supply. • In absence of dietary carbohydrate, the needed glucose must be made from non- carbohydrate precursors. • That process is called gluconeogenesis. Slide 3 Gluconeogenesis, con’t. • Brain and muscle consume most of the glucose. • Liver and kidney are the main sites of gluconeogenesis. • Substrates include pyruvate, lactate, glycerol, most amino acids, and all TCA intermediates. • Fatty acids cannot be converted to glucose in animals. • (They can in plants because of the glyoxalate cycle.) Chapter 23, page 1 Slide 4 Remember it is necessary for the pathways to differ in some respects, Gluconeogenesis, con’t. so that the overall DG can be negative in each direction. Usually • Substrates include anything that can be converted the steps with large negative DG of to phosphoenolpyruvate . one pathway are replaced in the • Many of the reactions are the same as those in reverse pathway with reactions that glycolysis. have a large negative DG in the • All glycolytic reactions which are near equilibrium can opposite direction. operate in both directions. • The three glycolytic reactions far from equilibrium (large -DG) must be bypassed. • A side by side comparison is shown in Fig 23.1. Slide 5 Unique Reactions of Gluconeogenesis • Recall that pyruvate kinase, though named in reverse, is not reversible and has a DG of –23 kJ/mol. -

GC-MS Metabolic Profile and -Glucosidase-, -Amylase-, Lipase-, and Acetylcholinesterase-Inhibitory Activities of Eight Peach
molecules Article GC-MS Metabolic Profile and α-Glucosidase-, α-Amylase-, Lipase-, and Acetylcholinesterase-Inhibitory Activities of Eight Peach Varieties Dasha Mihaylova 1,* , Ivelina Desseva 2,* , Aneta Popova 3 , Ivayla Dincheva 4 , Radka Vrancheva 2 , Anna Lante 5 and Albert Krastanov 1 1 Department of Biotechnology, Technological Faculty, University of Food Technologies, 4002 Plovdiv, Bulgaria; [email protected] 2 Department of Analytical Chemistry and Physical Chemistry, Technological Faculty, University of Food Technologies, 4002 Plovdiv, Bulgaria; [email protected] 3 Department of Catering and Tourism, Economics Faculty, University of Food Technologies, 4002 Plovdiv, Bulgaria; [email protected] 4 AgroBioInstitute, Agricultural Academy, 8 Dr. Tsankov Blvd., 1164 Sofia, Bulgaria; [email protected] 5 Department of Agronomy, Food, Natural Resources, Animals, and Environment—DAFNAE, Agripolis, University of Padova, 35020 Legnaro, Italy; [email protected] * Correspondence: [email protected] (D.M.); [email protected] (I.D.) Abstract: The inhibition of certain digestive enzymes by target food matrices represents a new approach in the treatment of socially significant diseases. Proving the ability of fruits to inhibit such enzymes can support the inclusion of specific varieties in the daily diets of patients with diabetes, obesity, Alzheimer’s disease, etc., providing them with much more than just valuable Citation: Mihaylova, D.; Desseva, I.; micro- and macromolecules. The current study aimed atidentifying and comparing the GC-MS Popova, A.; Dincheva, I.; Vrancheva, metabolic profiles of eight peach varieties (“Filina”, “Ufo 4, “Gergana”, “Laskava”, “July Lady”, R.; Lante, A.; Krastanov, A. GC-MS “Flat Queen”, “Evmolpiya”, and “Morsiani 90”) grown in Bulgaria (local and introduced) and to Metabolic Profile and α-Glucosidase-, evaluate the inhibitory potential of their extracts towards α-glucosidase, α-amylase, lipase, and α-Amylase-, Lipase-, and acetylcholinesterase. -

The Role of Phospholipase D (Pld) and Grb2 in Chemotaxis
THE ROLE OF PHOSPHOLIPASE D (PLD) AND GRB2 IN CHEMOTAXIS A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science By KATIE J. KNAPEK B.S., Indiana University of Pennyslvania, 2006 2008 Wright State University WRIGHT STATE UNIVERSITY SCHOOL OF GRADUATE STUDIES December 19, 2008 I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPERVISON BY Katie J. Knapek ENTITLED The Role of Phospholipase D (PLD) and Grb2 in Chemotaxis BE ACCEPTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science. _____________________________ Julian Gomez-Cambronero, Ph. D. Thesis Director _____________________________ Barbara Hull, Ph. D. Program Director Committee on Final Examination _______________________ Julian Gomez-Cambronero, Ph. D. _______________________ Nancy Bigley, Ph. D. _______________________ Mill Miller, Ph. D. ________________________ Joseph F. Thomas Jr., Ph. D. Dean, School of Graduate Studies ABSTRACT Knapek, Katie J. M.S., Department of Biological Sciences, Microbiology and Immunology Program, Wright State University, 2008. The Role of Phospholipase D (PLD) and Grb2 in Chemotaxis. Phospholipase D (PLD) is an enzyme that hydrolyzes phosphatidylcholine yielding choline and phosphatidic acid. PLD is activated by mitogens (lead to cell division) and motogens (leading to cell migration). PLD is known to contribute to cellular proliferation and deregulated expression of PLD has been implicated in several human cancers. PLD has been found to play a role in leukocyte chemotaxis and adhesion as studied through the formation of chemokine gradients. We have established a model of cell migration comprising three cell lines: macrophages RAW 264.7 and LR-5 (for innate defense), and fibroblast COS-7 cells (for wound healing). -

Multiple Roles of Phosphoinositide-Specific Phospholipase C Isozymes
BMB reports Mini Review Multiple roles of phosphoinositide-specific phospholipase C isozymes Pann-Ghill Suh1,*, Jae-Il Park1, Lucia Manzoli2, Lucio Cocco2, Joanna C. Peak3, Matilda Katan3, Kiyoko Fukami4, Tohru Kataoka5, Sanguk Yun1 & Sung Ho Ryu1 1Division of Molecular and Life Sciences, Pohang University of Science and Technology, Pohang, Korea, 2Cellular Signaling Laboratory, Department of Anatomical Sciences, University of Bologna, Via Irnerio, 48 I-40126, Bologna, Italy, 3Cancer Research UK Centre for Cell and Molecular Biology, Chester Beatty Laboratories, The Institute of Cancer Research, Fulham Road, London SW3 6JB, UK, 4Laboratory of Genome and Biosignal, Tokyo University of Pharmacy and Life Science, 1432-1 Horinouchi, Hachioji, 192-0392 Tokyo, Japan, 5Division of Molecular Biology, Department of Biochemistry and Molecular Biology, Kobe University Graduate School of Medicine, Chuo-ku, Kobe, Japan Phosphoinositide-specific phospholipase C is an effector mole- cellular calcium release (1-3; Fig. 1a). cule in the signal transduction process. It generates two sec- The first evidence of PLC activity was suggested by Hokin et ond messengers, inositol-1,4,5-trisphosphate and diacylglycer- al. in 1953 who reported specific hydrolysis of phospholipids ol from phosphatidylinositol 4,5-bisphosphate. Currently, thir- in pigeon’s pancreas slices after cholinergic stimulation (4). teen mammal PLC isozymes have been identified, and they are The authors showed that the enhanced turnover of phosphor- divided into six groups: PLC-β, -γ, -δ, -ε, -ζ and -η. Sequence ylinositol groups of phosphatidylinositol occurred in cells as a analysis studies demonstrated that each isozyme has more response to a variety of stimuli. In 1983, Streb et al. -

Deliverable 1.5 Model of Plant Photosynthesis
PROJECT ACRONYM: FutureAgriculture PROJECT TITLE: Transforming the future of agriculture through synthetic photorespiration Deliverable 1.5 Model of plant photosynthesis TOPIC H2020-FETOPEN-2014-2015-RIA GA 686330 Arren Bar-Even CONTACT PERSON [email protected] NATURE Report (R) DISSEMINATION LEVEL PU DUE DATE 31/12/2018 ACTUAL DELIVERED DATE 14/12/2018 AUTHOR Christian Edlich-Muth This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 686330. Table of Revisions REVISION NUMBER DATE WORK PERFORMED CONTRIBUTOR(S) 1 05/12/2018 Document preparation MPIMP 2 06/12/2018 Formatting IN 3 13/12/2018 Revision Consortium D1.5 – Model of plant photosynthesis Page 2 03/04/2018 Contents 1ExecutiveSummary 5 2Cooperationbetweenparticipants 6 3 Core report 7 3.1 Theory............................................. 7 3.1.1 Pathways . 7 3.1.2 The stoichiometric consumer model of photosynthesis . 9 3.1.3 Stoichiometric-kineticmodel. 13 3.1.4 Physicochemicalparameters. 24 3.2 Results............................................. 27 3.2.1 Parametersandvariables .............................. 27 3.2.2 Rubiscokinetics ................................... 28 3.2.3 Limitation by light . 30 3.2.4 LimitationbyenzymeactivitiesotherthanRubisco. 31 3.2.5 Limitation by a Calvin cycle enzyme . 31 3.2.6 Limitationbyaphotorespiratoryenzyme . 34 3.2.7 Limitationbyaphotorespirationcarboxylase . 35 4 Conclusions 37 D1.5Modelofplantphotosynthesis Page3 03/04/2018 Table of Charts 1 Bypass coefficients ........................................ 11 2 Flux distribtion of the Calvin cycle and photorespiratory bypasses . 20 3 Kinetic constants of Rubisco and carbon dioxide diffusion .................. 28 4 Calvin cycle enzyme total activities . 33 Table of Figures 1 Energy requirements of native photorespiration and the Calvin cycle . -

Pancreatic Amylase and Lipase Plasma Concentrations Are
Diabetes Care Volume 38, May 2015 e71 Pancreatic Amylase and Lipase Plasma David P. Sonne,1 Tina Vilsbøll,1 and Filip K. Knop1,2 Concentrations Are Unaffected by Increments in Endogenous GLP-1 Levels Following Liquid Meal Tests Diabetes Care 2015;38:e71–e72 | DOI: 10.2337/dc14-2751 Recent findings have suggested that amylase and lipase in plasma following patients with type 2 diabetes (P , incretin-based therapies promote pan- the ingestion of oral glucose and three 0.05), whereas lipase concentrations creatic inflammation and possibly cell isocaloric and isovolemic liquid mealsd were similar. Neither of the enzymes proliferation within the endocrine and all of which exerted normal endogenous increased following nutrient ingestion, exocrine pancreas (1). However, these GLP-1 secretion (4)din patients with suggesting that postprandial elevations studies have been met with substan- type 2 diabetes and matched control of endogenous GLP-1 (two- to three- tive criticism based on technical and subjects. fold) cannot trigger enzyme release methodical issues (2). Nevertheless, Detailed description of the experimen- from the human pancreas, at least not incretin-based therapies seem to result tal procedures and subjects was provided acutely. in small increases (within normal range) previously (4). In short, pancreas-specific These results suggest that the obser- in plasma concentrations of amylase amylase and lipase concentrations were vation of elevated plasma amylase and andlipaseinpatientsreceivingthese measured in plasma from 15 patients lipase -

Functional Role of Pentose Phosphate Pathway and Glutamine in Cancer Cell Metabolism Ibrahim Halil Polat
Functional role of pentose phosphate pathway and glutamine in cancer cell metabolism Ibrahim Halil Polat To cite this version: Ibrahim Halil Polat. Functional role of pentose phosphate pathway and glutamine in cancer cell metabolism. Human health and pathology. Universitat de Barcelona, 2016. English. NNT : 2016GREAS031. tel-01722703 HAL Id: tel-01722703 https://tel.archives-ouvertes.fr/tel-01722703 Submitted on 5 Mar 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour obtenir le grade de DOCTEUR DE LA COMMUNAUTÉ UNIVERSITÉ GRENOBLE ALPES préparée dans le cadre d’une cotutelle entre la Communauté Université Grenoble Alpes et Université de Barcelona Spécialité : BIS-Biotechnologie, instrumentation, signal et imagerie pour la biologie, la médecine et l’environnement Arrêté ministériel : 25 mai 2016 Présentée par « Ibrahim Halil Polat » Thèse dirigée par « Philippe Sabatier » codirigée par « Marta Cascante » préparée au sein des Laboratoires « TIMC-IMAG » et « BQI » dans les Écoles Doctorales « EDISCE » et « Ecole Doctorale de l’Université de Barcelone » Rôle fonctionnel des pentoses phosphates et glutamine dans le métabolisme des cellules cancéreuses Thèse soutenue publiquement le « 4 novembre 2016 », devant le jury composé de : M. Santiago Imperial Rodenas Professeur à l’Université de Barcelone (Président) Mme Loranne Agius Professeur à l’Université de Newcastle Upon Tyne (Rapporteur) M.