RESEARCH ARTICLE Regulatory Network of Micrornas, Target
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Global Characteristics of Csig-Associated Gene Expression Changes in Human Hek293 Cells and the Implications for Csig Regulating Cell Proliferation and Senescence
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Frontiers - Publisher Connector ORIGINAL RESEARCH published: 15 May 2015 doi: 10.3389/fendo.2015.00069 Global characteristics of CSIG-associated gene expression changes in human HEK293 cells and the implications for CSIG regulating cell proliferation and senescence Liwei Ma, Wenting Zhao, Feng Zhu, Fuwen Yuan, Nan Xie, Tingting Li, Pingzhang Wang and Tanjun Tong* Research Center on Aging. Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Peking University Health Science Center, Beijing Key Laboratory of Protein Posttranslational Modifications and Cell Function, Edited by: Beijing, China Wen Zhou, Columbia University, USA Reviewed by: Cellular senescence-inhibited gene (CSIG), also named as ribosomal_L1 domain-contain- Jian Zhong, ing 1 (RSL1D1), is implicated in various processes including cell cycle regulation, cellular Mayo Clinic, USA Xiaoxu Zheng, senescence, apoptosis, and tumor metastasis. However, little is known about the regulatory University of Maryland Baltimore, USA mechanism underlying its functions. To screen important targets and signaling pathways Wensi Tao, University of Miami, USA modulated by CSIG, we compared the gene expression profiles in CSIG-silencing and control *Correspondence: HEK293 cells using Affymetrix microarray Human Genome U133 Plus 2.0 GeneChips. A Tanjun Tong, total of 590 genes displayed statistically significant expression changes, with 279 genes Department of Biochemistry and up-regulated and 311 down-regulated, respectively. These genes are involved in a broad array Molecular Biology, Research Center on Aging, Peking University Health of biological processes, mainly in transcriptional regulation, cell cycle, signal transduction, Science Center, 38 Xueyuan Road, oxidation reduction, development, and cell adhesion. -

Supplementary Table S1. Upregulated Genes Differentially
Supplementary Table S1. Upregulated genes differentially expressed in athletes (p < 0.05 and 1.3-fold change) Gene Symbol p Value Fold Change 221051_s_at NMRK2 0.01 2.38 236518_at CCDC183 0.00 2.05 218804_at ANO1 0.00 2.05 234675_x_at 0.01 2.02 207076_s_at ASS1 0.00 1.85 209135_at ASPH 0.02 1.81 228434_at BTNL9 0.03 1.81 229985_at BTNL9 0.01 1.79 215795_at MYH7B 0.01 1.78 217979_at TSPAN13 0.01 1.77 230992_at BTNL9 0.01 1.75 226884_at LRRN1 0.03 1.74 220039_s_at CDKAL1 0.01 1.73 236520_at 0.02 1.72 219895_at TMEM255A 0.04 1.72 201030_x_at LDHB 0.00 1.69 233824_at 0.00 1.69 232257_s_at 0.05 1.67 236359_at SCN4B 0.04 1.64 242868_at 0.00 1.63 1557286_at 0.01 1.63 202780_at OXCT1 0.01 1.63 1556542_a_at 0.04 1.63 209992_at PFKFB2 0.04 1.63 205247_at NOTCH4 0.01 1.62 1554182_at TRIM73///TRIM74 0.00 1.61 232892_at MIR1-1HG 0.02 1.61 204726_at CDH13 0.01 1.6 1561167_at 0.01 1.6 1565821_at 0.01 1.6 210169_at SEC14L5 0.01 1.6 236963_at 0.02 1.6 1552880_at SEC16B 0.02 1.6 235228_at CCDC85A 0.02 1.6 1568623_a_at SLC35E4 0.00 1.59 204844_at ENPEP 0.00 1.59 1552256_a_at SCARB1 0.02 1.59 1557283_a_at ZNF519 0.02 1.59 1557293_at LINC00969 0.03 1.59 231644_at 0.01 1.58 228115_at GAREM1 0.01 1.58 223687_s_at LY6K 0.02 1.58 231779_at IRAK2 0.03 1.58 243332_at LOC105379610 0.04 1.58 232118_at 0.01 1.57 203423_at RBP1 0.02 1.57 AMY1A///AMY1B///AMY1C///AMY2A///AMY2B// 208498_s_at 0.03 1.57 /AMYP1 237154_at LOC101930114 0.00 1.56 1559691_at 0.01 1.56 243481_at RHOJ 0.03 1.56 238834_at MYLK3 0.01 1.55 213438_at NFASC 0.02 1.55 242290_at TACC1 0.04 1.55 ANKRD20A1///ANKRD20A12P///ANKRD20A2/// -

S41467-020-18249-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-18249-3 OPEN Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain Lei Zhao1,2,17, Zhongqi Li 1,2,17, Joaquim S. L. Vong2,3,17, Xinyi Chen1,2, Hei-Ming Lai1,2,4,5,6, Leo Y. C. Yan1,2, Junzhe Huang1,2, Samuel K. H. Sy1,2,7, Xiaoyu Tian 8, Yu Huang 8, Ho Yin Edwin Chan5,9, Hon-Cheong So6,8, ✉ ✉ Wai-Lung Ng 10, Yamei Tang11, Wei-Jye Lin12,13, Vincent C. T. Mok1,5,6,14,15 &HoKo 1,2,4,5,6,8,14,16 1234567890():,; The molecular signatures of cells in the brain have been revealed in unprecedented detail, yet the ageing-associated genome-wide expression changes that may contribute to neurovas- cular dysfunction in neurodegenerative diseases remain elusive. Here, we report zonation- dependent transcriptomic changes in aged mouse brain endothelial cells (ECs), which pro- minently implicate altered immune/cytokine signaling in ECs of all vascular segments, and functional changes impacting the blood–brain barrier (BBB) and glucose/energy metabolism especially in capillary ECs (capECs). An overrepresentation of Alzheimer disease (AD) GWAS genes is evident among the human orthologs of the differentially expressed genes of aged capECs, while comparative analysis revealed a subset of concordantly downregulated, functionally important genes in human AD brains. Treatment with exenatide, a glucagon-like peptide-1 receptor agonist, strongly reverses aged mouse brain EC transcriptomic changes and BBB leakage, with associated attenuation of microglial priming. We thus revealed tran- scriptomic alterations underlying brain EC ageing that are complex yet pharmacologically reversible. -

Epigenetic Regulation of DNA Repair Genes and Implications for Tumor Therapy ⁎ ⁎ Markus Christmann , Bernd Kaina
Mutation Research-Reviews in Mutation Research xxx (xxxx) xxx–xxx Contents lists available at ScienceDirect Mutation Research-Reviews in Mutation Research journal homepage: www.elsevier.com/locate/mutrev Review Epigenetic regulation of DNA repair genes and implications for tumor therapy ⁎ ⁎ Markus Christmann , Bernd Kaina Department of Toxicology, University of Mainz, Obere Zahlbacher Str. 67, D-55131 Mainz, Germany ARTICLE INFO ABSTRACT Keywords: DNA repair represents the first barrier against genotoxic stress causing metabolic changes, inflammation and DNA repair cancer. Besides its role in preventing cancer, DNA repair needs also to be considered during cancer treatment Genotoxic stress with radiation and DNA damaging drugs as it impacts therapy outcome. The DNA repair capacity is mainly Epigenetic silencing governed by the expression level of repair genes. Alterations in the expression of repair genes can occur due to tumor formation mutations in their coding or promoter region, changes in the expression of transcription factors activating or Cancer therapy repressing these genes, and/or epigenetic factors changing histone modifications and CpG promoter methylation MGMT Promoter methylation or demethylation levels. In this review we provide an overview on the epigenetic regulation of DNA repair genes. GADD45 We summarize the mechanisms underlying CpG methylation and demethylation, with de novo methyl- TET transferases and DNA repair involved in gain and loss of CpG methylation, respectively. We discuss the role of p53 components of the DNA damage response, p53, PARP-1 and GADD45a on the regulation of the DNA (cytosine-5)- methyltransferase DNMT1, the key enzyme responsible for gene silencing. We stress the relevance of epigenetic silencing of DNA repair genes for tumor formation and tumor therapy. -

The Emerging Role of Cohesin in the DNA Damage Response
G C A T T A C G G C A T genes Review The Emerging Role of Cohesin in the DNA Damage Response Ireneusz Litwin * , Ewa Pilarczyk and Robert Wysocki Institute of Experimental Biology, University of Wroclaw, 50-328 Wroclaw, Poland; [email protected] (E.P.); [email protected] (R.W.) * Correspondence: [email protected]; Tel.: +48-71-375-4126 Received: 29 October 2018; Accepted: 21 November 2018; Published: 28 November 2018 Abstract: Faithful transmission of genetic material is crucial for all organisms since changes in genetic information may result in genomic instability that causes developmental disorders and cancers. Thus, understanding the mechanisms that preserve genome integrity is of fundamental importance. Cohesin is a multiprotein complex whose canonical function is to hold sister chromatids together from S-phase until the onset of anaphase to ensure the equal division of chromosomes. However, recent research points to a crucial function of cohesin in the DNA damage response (DDR). In this review, we summarize recent advances in the understanding of cohesin function in DNA damage signaling and repair. First, we focus on cohesin architecture and molecular mechanisms that govern sister chromatid cohesion. Next, we briefly characterize the main DDR pathways. Finally, we describe mechanisms that determine cohesin accumulation at DNA damage sites and discuss possible roles of cohesin in DDR. Keywords: cohesin; cohesin loader; DNA double-strand breaks; replication stress; DNA damage tolerance 1. Introduction Genomes of all living organisms are continuously challenged by endogenous and exogenous insults that threaten genome stability. It has been estimated that human cells suffer more than 70,000 DNA lesions per day, most of which are single-strand DNA breaks (SSBs) [1]. -

Involvement of MBD4 Inactivation in Mismatch Repair-Deficient Tumorigenesis
Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Tricarico, R., S. Cortellino, A. Riccio, S. Jagmohan-Changur, H. van der Klift, J. Wijnen, D. Turner, et al. 2015. “Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis.” Oncotarget 6 (40): 42892-42904. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:26318553 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA www.impactjournals.com/oncotarget/ Oncotarget, Vol. 6, No. 40 Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis Rossella Tricarico1, Salvatore Cortellino2, Antonio Riccio3, Shantie Jagmohan- Changur4, Heleen Van der Klift5, Juul Wijnen5, David Turner6, Andrea Ventura7, Valentina Rovella8, Antonio Percesepe9, Emanuela Lucci-Cordisco10, Paolo Radice11, Lucio Bertario11, Monica Pedroni12, Maurizio Ponz de Leon12, Pietro Mancuso1,13, Karthik Devarajan14, Kathy Q. Cai15, Andres J.P. Klein-Szanto15, Giovanni Neri10, Pål Møller16, Alessandra Viel17, Maurizio Genuardi10, Riccardo Fodde4, Alfonso Bellacosa1 1Cancer Epigenetics and Cancer Biology Programs, Fox Chase Cancer Center, Philadelphia, Pennsylvania, United States 2IFOM-FIRC Institute of Molecular Oncology, Milan, -

Mismatch Repair
Downloaded from genesdev.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair Gerald T. Marsischky, Nicole Filosi, Michael F. Kane, and Richard Kolodner I Charles A. Dana Division of Human Cancer Genetics, Dana-Farber Cancer Institute, Boston, Massachusetts 09.115 USA; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115 USA Saccharomyces cerevisiae encodes six genes, MSH1-6, which encode proteins related to the bacterial MutS protein. In this study the role of MSH2, MSH3, and MSH6 in mismatch repair has been examined by measuring the rate of accumulating mutations and mutation spectrum in strains containing different combinations of rash2, rash3, and rash6 mutations and by studying the physical interaction between the MSH2 protein and the MSH3 and MSH6 proteins. The results indicate that S. cerevisiae has two pathways of MSH2-dependent mismatch repair: one that recognizes single-base mispairs and requires MSH2 and MSH6, and a second that recognizes insertion/deletion mispairs and requires a combination of either MSH2 and MSH6 or MSH2 and MSH3. The redundancy of MSH3 and MSH6 explains the greater prevalence of brash2 mutations in HNPCC families and suggests how the role of brash3 and hmsh6 mutations in cancer susceptibility could be analyzed. [Key Words: Cancer; mutagenesis; mismatch repair; routS; MSH2; MSH3; MSH6; Saccharomyces cerevisiae] Received October 17, 1995; revised version accepted January 17, 1996. DNA mismatch repair plays a number of roles in the cell al. 1993; Umar et al. 1994; Boyer et al. -

Original Article Rnai Screening Identifies KAT8 As a Key Molecule Important for Cancer Cell Survival
Int J Clin Exp Pathol 2013;6(5):870-877 www.ijcep.com /ISSN:1936-2625/IJCEP1302018 Original Article RNAi screening identifies KAT8 as a key molecule important for cancer cell survival Shuang Zhang1,2, Xianhong Liu2, Yong Zhang1, Ying Cheng2*, Yang Li1* 1Department of Pathophysiology, Norman Bathune College of Medical Sciences, Jilin University, Changchun, China; 2The first Department of Internal Medicine, Jilin Province Cancer Hospital, Changchun, China. *Equal contri- bution. Received February 16, 2013; Accepted April 1, 2013; Epub April 15, 2013; Published April 30, 2013 Abstract: Histone acetyltransferases (HATs) regulate many critical cancer events, including transcriptional regula- tion of oncogene and tumor suppressors, chromatin structure and DNA damage response. Abnormal expression of HATs has been reported in a number of cancers. However, cellular functions of HATs in cancer and molecular mechanisms remain largely unclear. Here, we performed a lentiviral vector-mediated RNAi screen to systematically address the function of HATs in lung cancer cell growth and viability. We identified 8 HATs genes involved in A549 cell viability. Further experiments showed that KAT8 regulates G2/M cell cycle arrest through AKT/ERK-cyclin D1 signaling. Moreover, KAT8 inhibition led to p53 induction and subsequently reduced bcl-2 expression. Our results demonstrate an important role of KAT8 in cancer and suggest that KAT8 could be a novel cancer therapeutic target. Keywords: HATs, RNAi screen, KAT8, cyclin D1, p53, cell survival Introduction well as DNA replication. Moreover, HATs gene expression profiles have been reported to be Histone acetylation, one of the most extensive- associated with pathological and clinical out- ly characterized epigenetic modifications, is comes in breast cancer [6]. -

The Properties of Msh2–Msh6 ATP Binding Mutants Suggest a Signal
ARTICLE cro Author’s Choice The properties of Msh2–Msh6 ATP binding mutants suggest a signal amplification mechanism in DNA mismatch repair Received for publication, August 19, 2018, and in revised form, September 17, 2018 Published, Papers in Press, September 20, 2018, DOI 10.1074/jbc.RA118.005439 William J. Graham, V‡, Christopher D. Putnam‡§, and X Richard D. Kolodner‡¶ʈ**1 From the ‡Ludwig Institute for Cancer Research San Diego, Departments of §Medicine and ¶Cellular and Molecular Medicine, ʈMoores-UCSD Cancer Center, and **Institute of Genomic Medicine, University of California School of Medicine, San Diego, La Jolla, California 92093-0669 Edited by Patrick Sung DNA mismatch repair (MMR) corrects mispaired DNA bases synthesis (1–6). MMR also acts on some forms of chemically and small insertion/deletion loops generated by DNA replica- damaged DNA bases as well as on mispairs present in hetero- tion errors. After binding a mispair, the eukaryotic mispair rec- duplex DNA intermediates formed during recombination ognition complex Msh2–Msh6 binds ATP in both of its nucle- (1–6). MMR proteins also act in a pathway that suppresses otide-binding sites, which induces a conformational change recombination between homologous but divergent DNAs resulting in the formation of an Msh2–Msh6 sliding clamp that (1–6) and function in a DNA damage response pathway for releases from the mispair and slides freely along the DNA. How- different types of DNA-damaging agents, including some che- ever, the roles that Msh2–Msh6 sliding clamps play in MMR motherapeutic agents (2). As a result of the role that MMR plays remain poorly understood. -

Comprehensive Biological Information Analysis of PTEN Gene in Pan-Cancer
Comprehensive biological information analysis of PTEN gene in pan-cancer Hang Zhang Shanghai Medical University: Fudan University https://orcid.org/0000-0002-5853-7754 Wenhan Zhou Shanghai Medical University: Fudan University Xiaoyi Yang Shanghai Jiao Tong University School of Medicine Shuzhan Wen Shanghai Medical University: Fudan University Baicheng Zhao Shanghai Medical University: Fudan University Jiale Feng Shanghai Medical University: Fudan University Shuying Chen ( [email protected] ) https://orcid.org/0000-0002-9215-9777 Primary research Keywords: PTEN, correlated genes, TCGA, GEPIA, UALCAN, GTEx, expression, cancer Posted Date: April 12th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-388887/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/21 Abstract Background PTEN is a multifunctional tumor suppressor gene mutating at high frequency in a variety of cancers. However, its expression in pan-cancer, correlated genes, survival prognosis, and regulatory pathways are not completely described. Here, we aimed to conduct a comprehensive analysis from the above perspectives in order to provide reference for clinical application. Methods we studied the expression levels in cancers by using data from TCGA and GTEx database. Obtain expression box plot from UALCAN database. Perform mutation analysis on the cBioportal website. Obtain correlation genes on the GEPIA website. Construct protein network and perform KEGG and GO enrichment analysis on the STRING database. Perform prognostic analysis on the Kaplan-Meier Plotter website. We also performed transcription factor prediction on the PROMO database and performed RNA-RNA association and RNA-protein interaction on the RNAup Web server and RPISEq. -
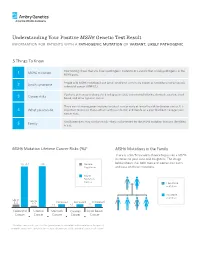
MSH6 Genetic Test Result Information for Patients with a Pathogenic Mutation Or Variant, Likely Pathogenic
Understanding Your Positive MSH6 Genetic Test Result information for patients with a pathogenic mutation or variant, likely pathogenic 5 Things To Know Your testing shows that you have a pathogenic mutation or a variant that is likely pathogenic in the 1 MSH6 mutation MSH6 gene. People with MSH6 mutations have Lynch syndrome, previously known as hereditary non-polyposis 2 Lynch syndrome colorectal cancer (HNPCC). You have an increased chance to develop colorectal, endometrial/uterine, stomach, ovarian, small 3 Cancer risks bowel, and other types of cancer. There are risk management options to detect cancer early or lower the risk to develop cancer. It is 4 What you can do important to discuss these options with your doctor, and decide on a plan that best manages your cancer risks. Family members may also be at risk – they can be tested for the MSH6 mutation that was identified 5 Family in you. MSH6 Mutation Lifetime Cancer Risks (%)* MSH6 Mutations in the Family There is a 50/50 random chance to pass on a MSH6 mutation to your sons and daughters. The image 20-44 ~44 General below shows that both men and women can carry Population and pass on these mutations. MSH6 Mutation Carrier Has MSH6 mutation No MSH6 mutation Up to Up to 5.5 Increased Increased Increased 2.7 <1 <1 <1 Colorectal Uterine Stomach Ovarian Small Bowel Cancer Cancer Cancer Cancer Cancer *The above cancer risks represent the typical range for individuals with a mutation in this gene. If available, cancer risks specific to the mutation found in you will be provided in your results report. -

Lynch Syndrome (MLH1, MSH2, MSH6, PMS2, EPCAM) - Update 2012
European Journal of Human Genetics (2013) 21; doi:10.1038/ejhg.2012.164 & 2013 Macmillan Publishers Limited All rights reserved 1018-4813/13 www.nature.com/ejhg CLINICAL UTILITY GENE CARD UPDATE Clinical utility gene card for: Lynch syndrome (MLH1, MSH2, MSH6, PMS2, EPCAM) - update 2012 Nils Rahner*,1, Verena Steinke2, Brigitte Schlegelberger3, Francois Eisinger4, Pierre Hutter5 and Sylviane Olschwang4 European Journal of Human Genetics (2013) 21, doi:10.1038/ejhg.2012.164; published online 15 August 2012 Update to: European Journal of Human Genetics (2010) 18, 1069; doi:10.1038/ejhg.2009.232; published online 27 January 2010 1. DISEASE CHARACTERISTICS MLH1;lossofMSH2/MSH6–analysisofMSH2; isolated loss of 1.1 Name of the disease (synonyms) MSH6 – analysis of MSH6; isolated loss of PMS2 – analysis of Lynch syndrome/HNPCC. PMS2). Germline analysis should include search for point mutations and large genomic deletions/duplications/insertions 1.2 OMIM# of the disease (e.g., by pre-screening (DHPLC), direct sequencing on gDNA or 276300, 613244. cDNA level, MLPA including promoter regions and EPCAM gene, Southern blot analysis). Mutation analysis of PMS2 should include 1.3 Name of the analysed genes or DNA/chromosome segments the analysis on RNA.9 Germline MLH1 promoter methylation MLH1, MSH2, MSH6, PMS2, and EPCAM. characterisation by MSP, bisulphite pyrosequencing, or MLPA (useful for diagnostic purpose, not for predictive testing). 1.4 OMIM# of the gene(s) MLH1 (120436), MSH2 (609309), MSH6 (600678), PMS2 (600259), EPCAM (185535). 1.7 Analytical validation Confirmation of mutation in an independent biological sample of the 1.5 Mutational spectrum index case or an affected relative.