Mismatch Repair
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Epigenetic Regulation of DNA Repair Genes and Implications for Tumor Therapy ⁎ ⁎ Markus Christmann , Bernd Kaina
Mutation Research-Reviews in Mutation Research xxx (xxxx) xxx–xxx Contents lists available at ScienceDirect Mutation Research-Reviews in Mutation Research journal homepage: www.elsevier.com/locate/mutrev Review Epigenetic regulation of DNA repair genes and implications for tumor therapy ⁎ ⁎ Markus Christmann , Bernd Kaina Department of Toxicology, University of Mainz, Obere Zahlbacher Str. 67, D-55131 Mainz, Germany ARTICLE INFO ABSTRACT Keywords: DNA repair represents the first barrier against genotoxic stress causing metabolic changes, inflammation and DNA repair cancer. Besides its role in preventing cancer, DNA repair needs also to be considered during cancer treatment Genotoxic stress with radiation and DNA damaging drugs as it impacts therapy outcome. The DNA repair capacity is mainly Epigenetic silencing governed by the expression level of repair genes. Alterations in the expression of repair genes can occur due to tumor formation mutations in their coding or promoter region, changes in the expression of transcription factors activating or Cancer therapy repressing these genes, and/or epigenetic factors changing histone modifications and CpG promoter methylation MGMT Promoter methylation or demethylation levels. In this review we provide an overview on the epigenetic regulation of DNA repair genes. GADD45 We summarize the mechanisms underlying CpG methylation and demethylation, with de novo methyl- TET transferases and DNA repair involved in gain and loss of CpG methylation, respectively. We discuss the role of p53 components of the DNA damage response, p53, PARP-1 and GADD45a on the regulation of the DNA (cytosine-5)- methyltransferase DNMT1, the key enzyme responsible for gene silencing. We stress the relevance of epigenetic silencing of DNA repair genes for tumor formation and tumor therapy. -

Involvement of MBD4 Inactivation in Mismatch Repair-Deficient Tumorigenesis
Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Tricarico, R., S. Cortellino, A. Riccio, S. Jagmohan-Changur, H. van der Klift, J. Wijnen, D. Turner, et al. 2015. “Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis.” Oncotarget 6 (40): 42892-42904. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:26318553 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA www.impactjournals.com/oncotarget/ Oncotarget, Vol. 6, No. 40 Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis Rossella Tricarico1, Salvatore Cortellino2, Antonio Riccio3, Shantie Jagmohan- Changur4, Heleen Van der Klift5, Juul Wijnen5, David Turner6, Andrea Ventura7, Valentina Rovella8, Antonio Percesepe9, Emanuela Lucci-Cordisco10, Paolo Radice11, Lucio Bertario11, Monica Pedroni12, Maurizio Ponz de Leon12, Pietro Mancuso1,13, Karthik Devarajan14, Kathy Q. Cai15, Andres J.P. Klein-Szanto15, Giovanni Neri10, Pål Møller16, Alessandra Viel17, Maurizio Genuardi10, Riccardo Fodde4, Alfonso Bellacosa1 1Cancer Epigenetics and Cancer Biology Programs, Fox Chase Cancer Center, Philadelphia, Pennsylvania, United States 2IFOM-FIRC Institute of Molecular Oncology, Milan, -

RESEARCH ARTICLE Regulatory Network of Micrornas, Target
DOI:http://dx.doi.org/10.7314/APJCP.2015.16.2.475 Regulatory Network of MicroRNAs, Target Genes, Transcription Factors and Host Genes in Endometrial Cancer RESEARCH ARTICLE Regulatory Network of MicroRNAs, Target Genes, Transcription Factors and Host Genes in Endometrial Cancer Lu-Chen Xue1,3, Zhi-Wen Xu2,3*, Kun-Hao Wang2,3, Ning Wang2,3, Xiao-Xu Zhang1, Shang Wang2,3 Abstract Genes and microRNAs (miRNAs) have important roles in human oncology. However, most of the biological factors are reported in disperse form which makes it hard to discover the pathology. In this study, genes and miRNAs involved in human endometrial cancer(EC) were collected and formed into regulatory networks following their interactive relations, including miRNAs targeting genes, transcription factors (TFs) regulating miRNAs and miRNAs included in their host genes. Networks are constructed hierarchically at three levels: differentially expressed, related and global. Among the three, the differentially expressed network is the most important and fundamental network that contains the key genes and miRNAs in EC. The target genes, TFs and miRNAs are differentially expressed in EC so that any mutation in them may impact on EC development. Some key pathways in networks were highlighted to analyze how they interactively influence other factors and carcinogenesis. Upstream and downstream pathways of the differentially expressed genes and miRNAs were compared and analyzed. The purpose of this study was to partially reveal the deep regulatory mechanisms in EC using a new method that combines comprehensive genes and miRNAs together with their relationships. It may contribute to cancer prevention and gene therapy of EC. -

The Properties of Msh2–Msh6 ATP Binding Mutants Suggest a Signal
ARTICLE cro Author’s Choice The properties of Msh2–Msh6 ATP binding mutants suggest a signal amplification mechanism in DNA mismatch repair Received for publication, August 19, 2018, and in revised form, September 17, 2018 Published, Papers in Press, September 20, 2018, DOI 10.1074/jbc.RA118.005439 William J. Graham, V‡, Christopher D. Putnam‡§, and X Richard D. Kolodner‡¶ʈ**1 From the ‡Ludwig Institute for Cancer Research San Diego, Departments of §Medicine and ¶Cellular and Molecular Medicine, ʈMoores-UCSD Cancer Center, and **Institute of Genomic Medicine, University of California School of Medicine, San Diego, La Jolla, California 92093-0669 Edited by Patrick Sung DNA mismatch repair (MMR) corrects mispaired DNA bases synthesis (1–6). MMR also acts on some forms of chemically and small insertion/deletion loops generated by DNA replica- damaged DNA bases as well as on mispairs present in hetero- tion errors. After binding a mispair, the eukaryotic mispair rec- duplex DNA intermediates formed during recombination ognition complex Msh2–Msh6 binds ATP in both of its nucle- (1–6). MMR proteins also act in a pathway that suppresses otide-binding sites, which induces a conformational change recombination between homologous but divergent DNAs resulting in the formation of an Msh2–Msh6 sliding clamp that (1–6) and function in a DNA damage response pathway for releases from the mispair and slides freely along the DNA. How- different types of DNA-damaging agents, including some che- ever, the roles that Msh2–Msh6 sliding clamps play in MMR motherapeutic agents (2). As a result of the role that MMR plays remain poorly understood. -
MSH6 Genetic Test Result Information for Patients with a Pathogenic Mutation Or Variant, Likely Pathogenic
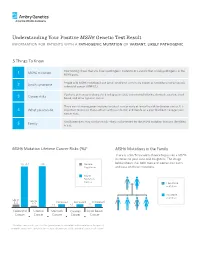
Understanding Your Positive MSH6 Genetic Test Result information for patients with a pathogenic mutation or variant, likely pathogenic 5 Things To Know Your testing shows that you have a pathogenic mutation or a variant that is likely pathogenic in the 1 MSH6 mutation MSH6 gene. People with MSH6 mutations have Lynch syndrome, previously known as hereditary non-polyposis 2 Lynch syndrome colorectal cancer (HNPCC). You have an increased chance to develop colorectal, endometrial/uterine, stomach, ovarian, small 3 Cancer risks bowel, and other types of cancer. There are risk management options to detect cancer early or lower the risk to develop cancer. It is 4 What you can do important to discuss these options with your doctor, and decide on a plan that best manages your cancer risks. Family members may also be at risk – they can be tested for the MSH6 mutation that was identified 5 Family in you. MSH6 Mutation Lifetime Cancer Risks (%)* MSH6 Mutations in the Family There is a 50/50 random chance to pass on a MSH6 mutation to your sons and daughters. The image 20-44 ~44 General below shows that both men and women can carry Population and pass on these mutations. MSH6 Mutation Carrier Has MSH6 mutation No MSH6 mutation Up to Up to 5.5 Increased Increased Increased 2.7 <1 <1 <1 Colorectal Uterine Stomach Ovarian Small Bowel Cancer Cancer Cancer Cancer Cancer *The above cancer risks represent the typical range for individuals with a mutation in this gene. If available, cancer risks specific to the mutation found in you will be provided in your results report. -

Lynch Syndrome (MLH1, MSH2, MSH6, PMS2, EPCAM) - Update 2012
European Journal of Human Genetics (2013) 21; doi:10.1038/ejhg.2012.164 & 2013 Macmillan Publishers Limited All rights reserved 1018-4813/13 www.nature.com/ejhg CLINICAL UTILITY GENE CARD UPDATE Clinical utility gene card for: Lynch syndrome (MLH1, MSH2, MSH6, PMS2, EPCAM) - update 2012 Nils Rahner*,1, Verena Steinke2, Brigitte Schlegelberger3, Francois Eisinger4, Pierre Hutter5 and Sylviane Olschwang4 European Journal of Human Genetics (2013) 21, doi:10.1038/ejhg.2012.164; published online 15 August 2012 Update to: European Journal of Human Genetics (2010) 18, 1069; doi:10.1038/ejhg.2009.232; published online 27 January 2010 1. DISEASE CHARACTERISTICS MLH1;lossofMSH2/MSH6–analysisofMSH2; isolated loss of 1.1 Name of the disease (synonyms) MSH6 – analysis of MSH6; isolated loss of PMS2 – analysis of Lynch syndrome/HNPCC. PMS2). Germline analysis should include search for point mutations and large genomic deletions/duplications/insertions 1.2 OMIM# of the disease (e.g., by pre-screening (DHPLC), direct sequencing on gDNA or 276300, 613244. cDNA level, MLPA including promoter regions and EPCAM gene, Southern blot analysis). Mutation analysis of PMS2 should include 1.3 Name of the analysed genes or DNA/chromosome segments the analysis on RNA.9 Germline MLH1 promoter methylation MLH1, MSH2, MSH6, PMS2, and EPCAM. characterisation by MSP, bisulphite pyrosequencing, or MLPA (useful for diagnostic purpose, not for predictive testing). 1.4 OMIM# of the gene(s) MLH1 (120436), MSH2 (609309), MSH6 (600678), PMS2 (600259), EPCAM (185535). 1.7 Analytical validation Confirmation of mutation in an independent biological sample of the 1.5 Mutational spectrum index case or an affected relative. -

Constitutional Mismatch Repair Deficiency Syndrome
Constitutional mismatch repair deficiency syndrome Description Constitutional mismatch repair deficiency (CMMRD) syndrome is a rare disorder that greatly increases the risk of developing one or more types of cancer in children and young adults. The cancers that most commonly occur in CMMRD syndrome are cancers of the colon (large intestine) and rectum (collectively referred to as colorectal cancer), brain, and blood (leukemia or lymphoma). Almost all people with CMMRD syndrome develop cancer before age 18, generally in late childhood. The age of diagnosis varies depending on the cancer type; brain cancers, leukemia, and lymphomas tend to occur at younger ages than colorectal cancer in people with CMMRD syndrome. It is estimated that 20 to 40 percent of people with CMMRD syndrome who develop cancer will develop another cancer later in life. People with CMMRD syndrome may develop multiple noncancerous (benign) growths ( adenomas) in the colon that are likely to become cancerous (malignant) over time. Brain cancers in CMMRD syndrome often involve certain cells called glial cells, causing gliomas or glioblastomas. The most common blood cancer in CMMRD syndrome is called non-Hodgkin lymphoma, which affects white blood cells. Other cancers that can occur in CMMRD syndrome include cancers of the small intestine, urinary tract, or uterine lining (endometrial cancer). Many people with CMMRD syndrome develop features similar to those that occur in a condition called neurofibromatosis type 1. These features include changes in skin coloring (pigmentation), which are characterized by one or more flat patches on the skin that are darker than the surrounding area (café-au-lait spots). Some affected individuals have freckling or patches of skin that are unusually light in color (hypopigmented). -

MSH6 and PMS2 Mutation Positive Australian Lynch Syndrome Families
Talseth-Palmer et al. Hereditary Cancer in Clinical Practice 2010, 8:5 http://www.hccpjournal.com/content/8/1/5 RESEARCH Open Access MSH6 and PMS2 mutation positive Australian Lynch syndrome families: novel mutations, cancer risk and age of diagnosis of colorectal cancer Bente A Talseth-Palmer1,2*, Mary McPhillips3, Claire Groombridge4, Allan Spigelman5, Rodney J Scott1,2,3 Abstract Background: Approximately 10% of Lynch syndrome families have a mutation in MSH6 and fewer families have a mutation in PMS2. It is assumed that the cancer incidence is the same in families with mutations in MSH6 as in families with mutations in MLH1/MSH2 but that the disease tends to occur later in life, little is known about families with PMS2 mutations. This study reports on our findings on mutation type, cancer risk and age of diagnosis in MSH6 and PMS2 families. Methods: A total of 78 participants (from 29 families) with a mutation in MSH6 and 7 participants (from 6 families) with a mutation in PMS2 were included in the current study. A database of de-identified patient information was analysed to extract all relevant information such as mutation type, cancer incidence, age of diagnosis and cancer type in this Lynch syndrome cohort. Cumulative lifetime risk was calculated utilising Kaplan-Meier survival analysis. Results: MSH6 and PMS2 mutations represent 10.3% and 1.9%, respectively, of the pathogenic mutations in our Australian Lynch syndrome families. We identified 26 different MSH6 and 4 different PMS2 mutations in the 35 families studied. We report 15 novel MSH6 and 1 novel PMS2 mutations. -

Co-Occurrence of Nonsense Mutations in MSH6 and MSH2 in Lynch Syndrome Families Evidencing That Not All Truncating Mutations Are Equal
Journal of Human Genetics (2016) 61, 151–156 & 2016 The Japan Society of Human Genetics All rights reserved 1434-5161/16 www.nature.com/jhg ORIGINAL ARTICLE Co-occurrence of nonsense mutations in MSH6 and MSH2 in Lynch syndrome families evidencing that not all truncating mutations are equal Carla Pinto1, Manuela Pinheiro1, Ana Peixoto1, Catarina Santos1, Isabel Veiga1, Patrícia Rocha1, Pedro Pinto1, Paula Lopes2, Manuela Baptista3, Rui Henrique2,4 and Manuel R Teixeira1,4 The majority of pathogenic mismatch repair (MMR) gene mutations detected in Lynch syndrome patients are truncating (frameshift or nonsense). However, the classification of terminal truncating mutations is sometimes difficult and predictive testing based on non-deleterious variants can have very serious consequences. Here, we report eight probands that have two germline nonsense mutations, namely MSH6 c.1030C4T, p.(Gln344Ter) and MSH2 c.2785C4T, p.(Arg929Ter), and one additional patient who presented only the MSH2 mutation previously reported as deleterious. The novel MSH6 truncating mutation was classified as deleterious, as it is predicted to encode a protein with loss of 1017 amino acid residues. The MSH2 mutation, which is expected to encode a protein lacking six amino acid residues, was considered a variant of unknown significance. Five tumors of the eight double-mutant individuals had normal MSH2 expression, whereas MSH6 immunoexpression was lost in all evaluable cases. None of the variants were detected in normal controls or associated with other MMR germline mutations in our series. This study emphasizes that not all truncating mutations are equal and that one must be cautious in the interpretation of the presumed deleterious effect of terminal frameshift or nonsense mutations. -

Immunohistochemistry for Microsatellite Instability Fact Sheet
Immunohistochemistry (IHC) for Microsatellite Instability Fact Sheet Frequently Asked Questions What is Lynch Syndrome? Lynch syndrome is a hereditary cancer syndrome associated with a significantly increased lifetime risk for colon, uterine, ovarian, stomach, and other cancers. If identified, patients can receive additional screening and prevention measures to help prevent cancer in the future. How is IHC useful in identifying patients with Lynch syndrome? IHC looks at the four mismatch repair proteins associated with Lynch syndrome. If these proteins are not present in the tumor, this means that the colon cancer could be due to Lynch syndrome. Why was this test done on my patient? Norton Hospital routinely performs IHC for Lynch syndrome on all colorectal resection specimens in order to maximize the identification of patients who are at increased risk for a second cancer and who have family members at significantly increased risk for cancer. Recent studies in the scientific literature have indicated this is the most effective way to identify patients with Lynch syndrome. This process has been approved by the Medical Executive Committee. How is this information helpful to me and my patient? Individuals with Lynch syndrome are at a significantly increased risk for developing cancer in the future. This information will be helpful in determining the ongoing management of your patient and may also aid in treatment decisions or eligibility for research studies. Patients can benefit from this information by understanding the cause of their cancer, their risk for subsequent cancers, and the risks for their family members. The family members of a patient can also be tested for Lynch syndrome, giving them the ability to make screening and prevention decisions that can prevent cancer in the future or allow it to be diagnosed at the earliest possible stage, when the cancer is most treatable. -

Mbd4 Inactivation Increases C3T Transition Mutations and Promotes Gastrointestinal Tumor Formation
Mbd4 inactivation increases C3T transition mutations and promotes gastrointestinal tumor formation Edmund Wong*, Kan Yang†‡, Mari Kuraguchi†‡, Uwe Werling*, Elena Avdievich*, Kunhua Fan†, Melissa Fazzari§, Bo Jin*, Anthony M. C. Brown†¶, Martin Lipkin†, and Winfried Edelmann*ʈ *Department of Cell Biology, §Biostatistics Core, Albert Einstein Cancer Center, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461; †Strang Cancer Research Laboratory at The Rockefeller University, New York, NY 10021; and ¶Department of Cell and Developmental Biology, Weill Medical College of Cornell University, New York, NY 10021 Communicated by Matthew D. Scharff, Albert Einstein College of Medicine, Bronx, NY, September 23, 2002 (received for review August 8, 2002) Mbd4 (methyl-CpG binding domain 4) is a novel mammalian repair tion that MBD4 contained an endonuclease activity that can enzyme that has been implicated biochemically in the repair of convert supercoiled plasmid DNA into nicked, linear molecules, mismatched G-T residues at methylated CpG sites. In addition, the it was suggested that MBD4 could function in mammalian MMR human protein has been shown to interact with the DNA mismatch in a manner reminiscent of the MutH endonuclease in bacterial repair protein MLH1. To clarify the role of Mbd4 in DNA repair in MMR (9). However, additional biochemical studies using dou- vivo and to examine the impact of Mbd4 inactivation on gastro- ble-stranded oligonucleotide substrates failed to detect an intestinal (GI) tumorigenesis, we introduced a null mutation into MBD4 endonuclease activity and therefore did not support a the murine Mbd4 gene by gene targeting. Heterozygous and role of MBD4 in mammalian MMR strand discrimination (7). -

The Rate of the Recurrent MSH6 Mutations in Ashkenazi Jewish Breast Cancer Patients
Cancer Causes & Control (2019) 30:97–101 https://doi.org/10.1007/s10552-018-1106-0 ORIGINAL PAPER The rate of the recurrent MSH6 mutations in Ashkenazi Jewish breast cancer patients Rinat Bernstein‑Molho1,3 · Yael Laitman2 · Hagit Schayek2 · Sarah Iomdin2 · Eitan Friedman2,3,4 Received: 12 August 2018 / Accepted: 26 November 2018 / Published online: 30 November 2018 © Springer Nature Switzerland AG 2018 Abstract Background Whether breast cancer (BC) should be considered within the spectrum of tumors in Lynch syndrome (LS) is unsettled. Recently, MSH6 and PMS2 germline mutations have reportedly been associated with an increased BC risk and with hereditary breast and ovarian cancer (HBOC) phenotype. We assessed the rates of the recurring Ashkenazi Jewish (AJ) mutations in the MSH6 gene (c.3984_3987dupGTCA and c.3959_3962delCAAG) in AJ cases with seemingly sporadic BC or HBOC phenotype, who were negative for the founder AJ BRCA1/2 mutations. Methods All AJ individuals, affected with BC ≤ 70 years and/or ovarian cancer at any age who were counseled, genotyped and tested negative for the BRCA1/2 founder mutations between January 2010 and February 2018 at the Oncogenetics unit, Sheba Medical Center, were genotyped for the AJ mutations in MSH6. Results Of 1016 genotyped participants (815 BC cases, 132 ovarian cancer cases, and 69 with more than one cancer), five carriers (0.49%) of the recurring AJ mutations in MSH6 were identified. All had BC, and two had personal history of addi- tional cancers (pancreatic, endometrial, colorectal). The rate of MSH6 mutations was 0.93% (4/429) when considering only cases with a personal or first-degree relative with LS-related cancer, and 0.17% (1/587) of cases with second-degree relative or no family history of LS-related cancers (p = 0.087).