Lynch Syndrome (Ls) Patient Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Dissertation Entitled the Role of Base Excision Repair And
A Dissertation Entitled The Role of Base Excision Repair and Mismatch Repair Proteins in the Processing of Cisplatin Interstrand Cross-Links. by Akshada Sawant Submitted to the Graduate Faculty as partial fulfillment of the requirements for the Doctor of Philosophy Degree in Biomedical Science Dr. Stephan M. Patrick, Committee Chair Dr. Kandace Williams, Committee Member Dr. William Maltese, Committee Member Dr. Manohar Ratnam, Committee Member Dr. David Giovannucci, Committee Member Dr. Patricia R. Komuniecki, Dean College of Graduate Studies The University of Toledo August 2014 Copyright 2014, Akshada Sawant This document is copyrighted material. Under copyright law, no parts of this document may be reproduced without the expressed permission of the author. An Abstract of The Role of Base Excision Repair and Mismatch Repair Proteins in the Processing of Cisplatin Interstrand Cross-Links By Akshada Sawant Submitted to the Graduate Faculty as partial fulfillment of the requirements for the Doctor of Philosophy Degree in Biomedical Science The University of Toledo August 2014 Cisplatin is a well-known anticancer agent that forms a part of many combination chemotherapeutic treatments used against a variety of human cancers. Despite successful treatment, the development of resistance is the major limitation of the cisplatin based therapy. Base excision repair modulates cisplatin cytotoxicity. Moreover, mismatch repair deficiency gives rise to cisplatin resistance and leads to poor prognosis of the disease. Various models have been proposed to explain this low level of resistance caused due to loss of MMR proteins. In our previous studies, we have shown that BER processing of the cisplatin ICLs is mutagenic. Our studies showed that these mismatches lead to the activation and the recruitment of mismatch repair proteins. -

HEREDITARY CANCER PANELS Part I
Pathology and Laboratory Medicine Clinic Building, K6, Core Lab, E-655 2799 W. Grand Blvd. HEREDITARY CANCER PANELS Detroit, MI 48202 855.916.4DNA (4362) Part I- REQUISITION Required Patient Information Ordering Physician Information Name: _________________________________________________ Gender: M F Name: _____________________________________________________________ MRN: _________________________ DOB: _______MM / _______DD / _______YYYY Address: ___________________________________________________________ ICD10 Code(s): _________________/_________________/_________________ City: _______________________________ State: ________ Zip: __________ ICD-10 Codes are required for billing. When ordering tests for which reimbursement will be sought, order only those tests that are medically necessary for the diagnosis and treatment of the patient. Phone: _________________________ Fax: ___________________________ Billing & Collection Information NPI: _____________________________________ Patient Demographic/Billing/Insurance Form is required to be submitted with this form. Most genetic testing requires insurance prior authorization. Due to high insurance deductibles and member policy benefits, patients may elect to self-pay. Call for more information (855.916.4362) Bill Client or Institution Client Name: ______________________________________________________ Client Code/Number: _____________ Bill Insurance Prior authorization or reference number: __________________________________________ Patient Self-Pay Call for pricing and payment options Toll -

Mismatch Repair Gene PMS2: Disease-Causing Germline
[CANCER RESEARCH 64, 4721–4727, July 15, 2004] Mismatch Repair Gene PMS2: Disease-Causing Germline Mutations Are Frequent in Patients Whose Tumors Stain Negative for PMS2 Protein, but Paralogous Genes Obscure Mutation Detection and Interpretation Hidewaki Nakagawa,1 Janet C. Lockman,1 Wendy L. Frankel,2 Heather Hampel,1 Kelle Steenblock,3 Lawrence J. Burgart,3 Stephen N. Thibodeau,3 and Albert de la Chapelle1 1Division of Human Cancer Genetics, Comprehensive Cancer Center, and 2Department of Surgical Pathology, The Ohio State University, Columbus, Ohio; and 3Departments of Laboratory Medicine and Pathology, Mayo Clinic and Foundation, Rochester, Minnesota ABSTRACT derived from a family described by Trimbath et al. (3). In two additional cases, children with cancer were heterozygous for germline ␣ The MutL heterodimer formed by mismatch repair (MMR) proteins mutations and even showed widespread microsatellite instability in MLH1 and PMS2 is a major component of the MMR complex, yet normal tissues, but the evidence appeared to support the notion of mutations in the PMS2 gene are rare in the etiology of hereditary non- polyposis colorectal cancer. Evidence from five published cases suggested recessive inheritance (even though a second mutation was not found), that contrary to the Knudson principle, PMS2 mutations cause hereditary in that a parent who had the same mutation had no cancer (4, 5). The nonpolyposis colorectal cancer or Turcot syndrome only when they are fifth patient reported was heterozygous for a PMS2 germline muta- biallelic in the germline or abnormally expressed. As candidates for PMS2 tion, but clinical features were not described (6). mutations, we selected seven patients whose colon tumors stained negative The Knudson two-hit model (7) applies to the MLH1, MSH2, and for PMS2 and positive for MLH1 by immunohistochemistry. -

Epigenetic Regulation of DNA Repair Genes and Implications for Tumor Therapy ⁎ ⁎ Markus Christmann , Bernd Kaina
Mutation Research-Reviews in Mutation Research xxx (xxxx) xxx–xxx Contents lists available at ScienceDirect Mutation Research-Reviews in Mutation Research journal homepage: www.elsevier.com/locate/mutrev Review Epigenetic regulation of DNA repair genes and implications for tumor therapy ⁎ ⁎ Markus Christmann , Bernd Kaina Department of Toxicology, University of Mainz, Obere Zahlbacher Str. 67, D-55131 Mainz, Germany ARTICLE INFO ABSTRACT Keywords: DNA repair represents the first barrier against genotoxic stress causing metabolic changes, inflammation and DNA repair cancer. Besides its role in preventing cancer, DNA repair needs also to be considered during cancer treatment Genotoxic stress with radiation and DNA damaging drugs as it impacts therapy outcome. The DNA repair capacity is mainly Epigenetic silencing governed by the expression level of repair genes. Alterations in the expression of repair genes can occur due to tumor formation mutations in their coding or promoter region, changes in the expression of transcription factors activating or Cancer therapy repressing these genes, and/or epigenetic factors changing histone modifications and CpG promoter methylation MGMT Promoter methylation or demethylation levels. In this review we provide an overview on the epigenetic regulation of DNA repair genes. GADD45 We summarize the mechanisms underlying CpG methylation and demethylation, with de novo methyl- TET transferases and DNA repair involved in gain and loss of CpG methylation, respectively. We discuss the role of p53 components of the DNA damage response, p53, PARP-1 and GADD45a on the regulation of the DNA (cytosine-5)- methyltransferase DNMT1, the key enzyme responsible for gene silencing. We stress the relevance of epigenetic silencing of DNA repair genes for tumor formation and tumor therapy. -

The Diagnostic Value of DNA Repair Gene in Breast Cancer Recurrence and Metastasis
The Diagnostic Value of DNA Repair Gene in Breast Cancer Recurrence and Metastasis Yongxin Yang Southwest Medical University Xiabin Li Southwest Medical University Liyue Hao Southwest Medical University Deyong Jiang Centers for Disease Control and Prevention Bin Wu Southwest Medical University Tao He Southwest Medical University Yan Tang ( [email protected] ) Research Keywords: PARP1, XRCC4, ERCC1, Breast cancer Posted Date: June 25th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-36932/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/19 Abstract Background: DNA repair genes play a vital role in the treatment of many cancers, and DNA repair genes can be used in breast cancer recurrence and metastasis research. We found that the expression of DNA repair genes in breast cancer patients after recurrence and metastasis is abnormal, however, the clinical predictive signicance of DNA repair genes is still elusive. Methods: The nested case-control method was used in patients with breast cancer recurrence and metastasis after surgery (n=109) and patients without recurrence and metastasis after surgery (n=109). The proteins and mRNA of DNA repair genes were detected by immunohistochemistry and Real-time PCR respectively. Results: PARP1(OR=1.485, 95%CI:1.279~1.725, P<0.05), XRCC4(OR= 1.419, 95%CI:1.217~ 1.656, P<0.05) and ERCC1 (OR=1.181, 95%CI: 1.032~1.353, P<0.05) were risk factors for postoperative recurrence and metastasis of breast cancer. Therefore, we used the ROC -

Deficiency in DNA Mismatch Repair of Methylation Damage Is a Major
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.18.388108; this version posted November 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Deficiency in DNA mismatch repair of methylation damage is a major 2 mutational process in cancer 3 1 1 1 1 1,* 4 Hu Fang , Xiaoqiang Zhu , Jieun Oh , Jayne A. Barbour , Jason W. H. Wong 5 6 1School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, The University of 7 Hong Kong, Hong Kong Special Administrative Region 8 9 *Correspondence: [email protected] 10 11 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.18.388108; this version posted November 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Abstract 2 DNA mismatch repair (MMR) is essential for maintaining genome integrity with its 3 deficiency predisposing to cancer1. MMR is well known for its role in the post- 4 replicative repair of mismatched base pairs that escape proofreading by DNA 5 polymerases following cell division2. Yet, cancer genome sequencing has revealed that 6 MMR deficient cancers not only have high mutation burden but also harbour multiple 7 mutational signatures3, suggesting that MMR has pleotropic effects on DNA repair. The 8 mechanisms underlying these mutational signatures have remained unclear despite 9 studies using a range of in vitro4,5 and in vivo6 models of MMR deficiency. -

WRN Promoter Methylation Possibly Connects Mucinous Differentiation, Microsatellite Instability and Cpg Island Methylator Phenotype in Colorectal Cancer
Modern Pathology (2008) 21, 150–158 & 2008 USCAP, Inc All rights reserved 0893-3952/08 $30.00 www.modernpathology.org WRN promoter methylation possibly connects mucinous differentiation, microsatellite instability and CpG island methylator phenotype in colorectal cancer Takako Kawasaki1,2, Mutsuko Ohnishi2, Yuko Suemoto2, Gregory J Kirkner3, Zhiqian Liu2, Hiroyuki Yamamoto4, Massimo Loda1,2, Charles S Fuchs2,3 and Shuji Ogino1,2 1Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA; 2Department of Medical Oncology, Dana–Farber Cancer Institute, Boston, MA, USA; 3Channing Laboratory, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA and 4First Department of Internal Medicine, Sapporo Medical University, Sapporo, Japan Werner syndrome is a premature aging syndrome characterized by early onset of cancer and abnormal cellular metabolism of glycosaminoglycan. The WRN helicase plays an important role in the maintenance of telomere function. WRN promoter methylation and gene silencing are common in colorectal cancer with the CpG island methylator phenotype (CIMP), which is associated with microsatellite instability (MSI) and mucinous tumors. However, no study has examined the relationship between mucinous differentiation, WRN methylation, CIMP and MSI in colorectal cancer. Utilizing 903 population-based colorectal cancers and real-time PCR (MethyLight), we quantified DNA methylation in WRN and eight other promoters (CACNA1G, CDKN2A, CRABP1, IGF2, MLH1, NEUROG1, RUNX3 and SOCS1) known to be specific for CIMP. Supporting WRN as a good CIMP marker, WRN methylation was correlated well with CIMP-high diagnosis (Z6/8 methylated promoters), demonstrating 89% sensitivity and 81% specificity. WRN methylation was associated with the presence of any mucinous component and Z50% mucinous component (Po0.0001). -

Involvement of MBD4 Inactivation in Mismatch Repair-Deficient Tumorigenesis
Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Tricarico, R., S. Cortellino, A. Riccio, S. Jagmohan-Changur, H. van der Klift, J. Wijnen, D. Turner, et al. 2015. “Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis.” Oncotarget 6 (40): 42892-42904. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:26318553 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA www.impactjournals.com/oncotarget/ Oncotarget, Vol. 6, No. 40 Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis Rossella Tricarico1, Salvatore Cortellino2, Antonio Riccio3, Shantie Jagmohan- Changur4, Heleen Van der Klift5, Juul Wijnen5, David Turner6, Andrea Ventura7, Valentina Rovella8, Antonio Percesepe9, Emanuela Lucci-Cordisco10, Paolo Radice11, Lucio Bertario11, Monica Pedroni12, Maurizio Ponz de Leon12, Pietro Mancuso1,13, Karthik Devarajan14, Kathy Q. Cai15, Andres J.P. Klein-Szanto15, Giovanni Neri10, Pål Møller16, Alessandra Viel17, Maurizio Genuardi10, Riccardo Fodde4, Alfonso Bellacosa1 1Cancer Epigenetics and Cancer Biology Programs, Fox Chase Cancer Center, Philadelphia, Pennsylvania, United States 2IFOM-FIRC Institute of Molecular Oncology, Milan, -

Mismatch Repair
Downloaded from genesdev.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair Gerald T. Marsischky, Nicole Filosi, Michael F. Kane, and Richard Kolodner I Charles A. Dana Division of Human Cancer Genetics, Dana-Farber Cancer Institute, Boston, Massachusetts 09.115 USA; Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115 USA Saccharomyces cerevisiae encodes six genes, MSH1-6, which encode proteins related to the bacterial MutS protein. In this study the role of MSH2, MSH3, and MSH6 in mismatch repair has been examined by measuring the rate of accumulating mutations and mutation spectrum in strains containing different combinations of rash2, rash3, and rash6 mutations and by studying the physical interaction between the MSH2 protein and the MSH3 and MSH6 proteins. The results indicate that S. cerevisiae has two pathways of MSH2-dependent mismatch repair: one that recognizes single-base mispairs and requires MSH2 and MSH6, and a second that recognizes insertion/deletion mispairs and requires a combination of either MSH2 and MSH6 or MSH2 and MSH3. The redundancy of MSH3 and MSH6 explains the greater prevalence of brash2 mutations in HNPCC families and suggests how the role of brash3 and hmsh6 mutations in cancer susceptibility could be analyzed. [Key Words: Cancer; mutagenesis; mismatch repair; routS; MSH2; MSH3; MSH6; Saccharomyces cerevisiae] Received October 17, 1995; revised version accepted January 17, 1996. DNA mismatch repair plays a number of roles in the cell al. 1993; Umar et al. 1994; Boyer et al. -

Predisposition to Hematologic Malignancies in Patients With
LETTERS TO THE EDITOR carcinomas but no internal cancer by the age of 29 years Predisposition to hematologic malignancies in and 9 years, respectively. patients with xeroderma pigmentosum Case XP540BE . This patient had a highly unusual pres - entation of MPAL. She was diagnosed with XP at the age Germline predisposition is a contributing etiology of of 18 months with numerous lentigines on sun-exposed hematologic malignancies, especially in children and skin, when her family emigrated from Morocco to the young adults. Germline predisposition in myeloid neo - USA. The homozygous North African XPC founder muta - plasms was added to the World Health Organization tion was present. 10 She had her first skin cancer at the age 1 2016 classification, and current management recommen - of 8 years, and subsequently developed more than 40 cuta - dations emphasize the importance of screening appropri - neous basal and squamous cell carcinomas, one melanoma 2 ate patients. Rare syndromes of DNA repair defects can in situ , and one ocular surface squamous neoplasm. She 3 lead to myeloid and/or lymphoid neoplasms. Here, we was diagnosed with a multinodular goiter at the age of 9 describe our experience with hematologic neoplasms in years eight months, with several complex nodules leading the defective DNA repair syndrome, xeroderma pigmen - to removal of her thyroid gland. Histopathology showed tosum (XP), including myelodysplastic syndrome (MDS), multinodular adenomatous/papillary hyperplasia. At the secondary acute myeloid leukemia (AML), high-grade age of 19 years, she presented with night sweats, fatigue, lymphoma, and an extremely unusual presentation of and lymphadenopathy. Laboratory studies revealed pancy - mixed phenotype acute leukemia (MPAL) with B, T and topenia with hemoglobin 6.8 g/dL, platelet count myeloid blasts. -

RESEARCH ARTICLE Regulatory Network of Micrornas, Target
DOI:http://dx.doi.org/10.7314/APJCP.2015.16.2.475 Regulatory Network of MicroRNAs, Target Genes, Transcription Factors and Host Genes in Endometrial Cancer RESEARCH ARTICLE Regulatory Network of MicroRNAs, Target Genes, Transcription Factors and Host Genes in Endometrial Cancer Lu-Chen Xue1,3, Zhi-Wen Xu2,3*, Kun-Hao Wang2,3, Ning Wang2,3, Xiao-Xu Zhang1, Shang Wang2,3 Abstract Genes and microRNAs (miRNAs) have important roles in human oncology. However, most of the biological factors are reported in disperse form which makes it hard to discover the pathology. In this study, genes and miRNAs involved in human endometrial cancer(EC) were collected and formed into regulatory networks following their interactive relations, including miRNAs targeting genes, transcription factors (TFs) regulating miRNAs and miRNAs included in their host genes. Networks are constructed hierarchically at three levels: differentially expressed, related and global. Among the three, the differentially expressed network is the most important and fundamental network that contains the key genes and miRNAs in EC. The target genes, TFs and miRNAs are differentially expressed in EC so that any mutation in them may impact on EC development. Some key pathways in networks were highlighted to analyze how they interactively influence other factors and carcinogenesis. Upstream and downstream pathways of the differentially expressed genes and miRNAs were compared and analyzed. The purpose of this study was to partially reveal the deep regulatory mechanisms in EC using a new method that combines comprehensive genes and miRNAs together with their relationships. It may contribute to cancer prevention and gene therapy of EC. -
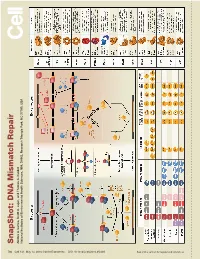
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases.