Somatic Variants of Potential Clinical Significance in the Tumors of BRCA
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

HEREDITARY CANCER PANELS Part I
Pathology and Laboratory Medicine Clinic Building, K6, Core Lab, E-655 2799 W. Grand Blvd. HEREDITARY CANCER PANELS Detroit, MI 48202 855.916.4DNA (4362) Part I- REQUISITION Required Patient Information Ordering Physician Information Name: _________________________________________________ Gender: M F Name: _____________________________________________________________ MRN: _________________________ DOB: _______MM / _______DD / _______YYYY Address: ___________________________________________________________ ICD10 Code(s): _________________/_________________/_________________ City: _______________________________ State: ________ Zip: __________ ICD-10 Codes are required for billing. When ordering tests for which reimbursement will be sought, order only those tests that are medically necessary for the diagnosis and treatment of the patient. Phone: _________________________ Fax: ___________________________ Billing & Collection Information NPI: _____________________________________ Patient Demographic/Billing/Insurance Form is required to be submitted with this form. Most genetic testing requires insurance prior authorization. Due to high insurance deductibles and member policy benefits, patients may elect to self-pay. Call for more information (855.916.4362) Bill Client or Institution Client Name: ______________________________________________________ Client Code/Number: _____________ Bill Insurance Prior authorization or reference number: __________________________________________ Patient Self-Pay Call for pricing and payment options Toll -

Mismatch Repair Gene PMS2: Disease-Causing Germline
[CANCER RESEARCH 64, 4721–4727, July 15, 2004] Mismatch Repair Gene PMS2: Disease-Causing Germline Mutations Are Frequent in Patients Whose Tumors Stain Negative for PMS2 Protein, but Paralogous Genes Obscure Mutation Detection and Interpretation Hidewaki Nakagawa,1 Janet C. Lockman,1 Wendy L. Frankel,2 Heather Hampel,1 Kelle Steenblock,3 Lawrence J. Burgart,3 Stephen N. Thibodeau,3 and Albert de la Chapelle1 1Division of Human Cancer Genetics, Comprehensive Cancer Center, and 2Department of Surgical Pathology, The Ohio State University, Columbus, Ohio; and 3Departments of Laboratory Medicine and Pathology, Mayo Clinic and Foundation, Rochester, Minnesota ABSTRACT derived from a family described by Trimbath et al. (3). In two additional cases, children with cancer were heterozygous for germline ␣ The MutL heterodimer formed by mismatch repair (MMR) proteins mutations and even showed widespread microsatellite instability in MLH1 and PMS2 is a major component of the MMR complex, yet normal tissues, but the evidence appeared to support the notion of mutations in the PMS2 gene are rare in the etiology of hereditary non- polyposis colorectal cancer. Evidence from five published cases suggested recessive inheritance (even though a second mutation was not found), that contrary to the Knudson principle, PMS2 mutations cause hereditary in that a parent who had the same mutation had no cancer (4, 5). The nonpolyposis colorectal cancer or Turcot syndrome only when they are fifth patient reported was heterozygous for a PMS2 germline muta- biallelic in the germline or abnormally expressed. As candidates for PMS2 tion, but clinical features were not described (6). mutations, we selected seven patients whose colon tumors stained negative The Knudson two-hit model (7) applies to the MLH1, MSH2, and for PMS2 and positive for MLH1 by immunohistochemistry. -

Deficiency in DNA Mismatch Repair of Methylation Damage Is a Major
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.18.388108; this version posted November 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Deficiency in DNA mismatch repair of methylation damage is a major 2 mutational process in cancer 3 1 1 1 1 1,* 4 Hu Fang , Xiaoqiang Zhu , Jieun Oh , Jayne A. Barbour , Jason W. H. Wong 5 6 1School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, The University of 7 Hong Kong, Hong Kong Special Administrative Region 8 9 *Correspondence: [email protected] 10 11 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.18.388108; this version posted November 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Abstract 2 DNA mismatch repair (MMR) is essential for maintaining genome integrity with its 3 deficiency predisposing to cancer1. MMR is well known for its role in the post- 4 replicative repair of mismatched base pairs that escape proofreading by DNA 5 polymerases following cell division2. Yet, cancer genome sequencing has revealed that 6 MMR deficient cancers not only have high mutation burden but also harbour multiple 7 mutational signatures3, suggesting that MMR has pleotropic effects on DNA repair. The 8 mechanisms underlying these mutational signatures have remained unclear despite 9 studies using a range of in vitro4,5 and in vivo6 models of MMR deficiency. -

Involvement of MBD4 Inactivation in Mismatch Repair-Deficient Tumorigenesis
Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Tricarico, R., S. Cortellino, A. Riccio, S. Jagmohan-Changur, H. van der Klift, J. Wijnen, D. Turner, et al. 2015. “Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis.” Oncotarget 6 (40): 42892-42904. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:26318553 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA www.impactjournals.com/oncotarget/ Oncotarget, Vol. 6, No. 40 Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis Rossella Tricarico1, Salvatore Cortellino2, Antonio Riccio3, Shantie Jagmohan- Changur4, Heleen Van der Klift5, Juul Wijnen5, David Turner6, Andrea Ventura7, Valentina Rovella8, Antonio Percesepe9, Emanuela Lucci-Cordisco10, Paolo Radice11, Lucio Bertario11, Monica Pedroni12, Maurizio Ponz de Leon12, Pietro Mancuso1,13, Karthik Devarajan14, Kathy Q. Cai15, Andres J.P. Klein-Szanto15, Giovanni Neri10, Pål Møller16, Alessandra Viel17, Maurizio Genuardi10, Riccardo Fodde4, Alfonso Bellacosa1 1Cancer Epigenetics and Cancer Biology Programs, Fox Chase Cancer Center, Philadelphia, Pennsylvania, United States 2IFOM-FIRC Institute of Molecular Oncology, Milan, -

Predisposition to Hematologic Malignancies in Patients With
LETTERS TO THE EDITOR carcinomas but no internal cancer by the age of 29 years Predisposition to hematologic malignancies in and 9 years, respectively. patients with xeroderma pigmentosum Case XP540BE . This patient had a highly unusual pres - entation of MPAL. She was diagnosed with XP at the age Germline predisposition is a contributing etiology of of 18 months with numerous lentigines on sun-exposed hematologic malignancies, especially in children and skin, when her family emigrated from Morocco to the young adults. Germline predisposition in myeloid neo - USA. The homozygous North African XPC founder muta - plasms was added to the World Health Organization tion was present. 10 She had her first skin cancer at the age 1 2016 classification, and current management recommen - of 8 years, and subsequently developed more than 40 cuta - dations emphasize the importance of screening appropri - neous basal and squamous cell carcinomas, one melanoma 2 ate patients. Rare syndromes of DNA repair defects can in situ , and one ocular surface squamous neoplasm. She 3 lead to myeloid and/or lymphoid neoplasms. Here, we was diagnosed with a multinodular goiter at the age of 9 describe our experience with hematologic neoplasms in years eight months, with several complex nodules leading the defective DNA repair syndrome, xeroderma pigmen - to removal of her thyroid gland. Histopathology showed tosum (XP), including myelodysplastic syndrome (MDS), multinodular adenomatous/papillary hyperplasia. At the secondary acute myeloid leukemia (AML), high-grade age of 19 years, she presented with night sweats, fatigue, lymphoma, and an extremely unusual presentation of and lymphadenopathy. Laboratory studies revealed pancy - mixed phenotype acute leukemia (MPAL) with B, T and topenia with hemoglobin 6.8 g/dL, platelet count myeloid blasts. -
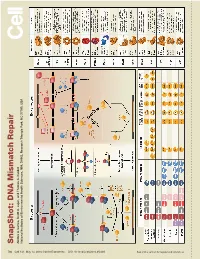
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

DNA Mismatch Repair Proteins MLH1 and PMS2 Can Be Imported to the Nucleus by a Classical Nuclear Import Pathway
Accepted Manuscript DNA mismatch repair proteins MLH1 and PMS2 can be imported to the nucleus by a classical nuclear import pathway Andrea C. de Barros, Agnes A.S. Takeda, Thiago R. Dreyer, Adrian Velazquez- Campoy, Boštjan Kobe, Marcos R.M. Fontes PII: S0300-9084(17)30308-5 DOI: 10.1016/j.biochi.2017.11.013 Reference: BIOCHI 5321 To appear in: Biochimie Received Date: 25 May 2017 Accepted Date: 22 November 2017 Please cite this article as: Keplinger M, Marhofer P, Moriggl B, Zeitlinger M, Muehleder-Matterey S, Marhofer D, Cutaneous innervation of the hand: clinical testing in volunteers shows that variability is far greater than claimed in textbooks, British Journal of Anaesthesia (2017), doi: 10.1016/j.bja.2017.09.008. This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. ACCEPTED MANUSCRIPT ABSTRACT MLH1 and PMS2 proteins form the MutL α heterodimer, which plays a major role in DNA mismatch repair (MMR) in humans. Mutations in MMR-related proteins are associated with cancer, especially with colon cancer. The N-terminal region of MutL α comprises the N-termini of PMS2 and MLH1 and, similarly, the C-terminal region of MutL α is composed by the C-termini of PMS2 and MLH1, and the two are connected by linker region. -

Male Factor Infertility and Health Karen Baker, MD Associate Professor Duke University, Division of Urology Fertility And…
Male Factor Infertility and Health Karen Baker, MD Associate Professor Duke University, Division of Urology Fertility and… • Cancer • Goals: • Heart disease • Review literature linking MFI to poor health • Metabolic syndrome • Diabetes • Discuss possible mechanisms common to MFI and cancer • Early death Cardiovascular risk • 3.5 million AARP members surveyed 1995-1996 • 137,903 men met criteria • 92% +paternity • mean age 62.7 yrs • 96.4% white • Age adjusted cardiovascular mortality risk 2.7/1000 person- years • Childless men 17%↑ cardiovascular mortality compared with men with + paternity Eisenberg, Hum Repro 2011 Cardiovascular risk • 3.5 million AARP members surveyed 1995-1996 • 137,903 men met criteria • 92% +paternity • mean age 62.7 yrs • 96.4% white • Age adjusted cardiovascular mortality risk 2.7/1000 person- years • Childless men 17%↑ cardiovascular mortality compared with men with + paternity Eisenberg, Hum Repro 2011 Chronic disease • Insurance claims 2001 - 2009 • 13,027 infertile men • Semen testing + MFI dx • 23,860 fertile men • Semen testing no MFI dx • 79,099 vasectomy • Age • Smoking by ICD9 • Obesity • Compared rates of common medical conditions • HTN, DM, peripheral vascular disease, cerebrovascular disease, ischemic heart by ICD9 disease, alcohol abuse, bipolar, etc. Chronic disease • ↑ medical disease in MFI vs Risk of developing chronic medical dz after dx MFI “semen testing” and vasectomies • DM HR 1.81 (95%CI 1.57-2.08) • Renal dz HR 1.6 (95%CI 1..14- 2.24) • Peripheral vascular dz HR 1.52 (95% CI 1.12-2.07) • -

Identification of the Mismatch Repair Genes PMS2 and MLH1 As P53 Target Genes by Using Serial Analysis of Binding Elements
Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements Jiguo Chen and Ivan Sadowski* Department of Biochemistry and Molecular Biology, University of British Columbia, 2146 Health Sciences Mall, Vancouver, BC, Canada V6T 1Z3 Edited by Mark T. Groudine, Fred Hutchinson Cancer Research Center, Seattle, WA, and approved February 22, 2005 (received for review September 23, 2004) The ability to determine the global location of transcription factor somes (11). Here, we describe a technology called serial analysis of binding sites in vivo is important for a comprehensive understand- binding elements (SABE) for globally identifying binding sites of ing of gene regulation in human cells. We have developed a mammalian transcription factors in vivo. SABE involves specific technology, called serial analysis of binding elements (SABE), ChIP (12), enrichment of ChIP DNA by using representational involving subtractive hybridization of chromatin immunoprecipi- difference analysis (13), and generation of sequence tags similar to tation-enriched DNA fragments followed by the generation and serial analysis of gene expression (SAGE) (14). By using this analysis of concatamerized sequence tags. We applied the SABE approach, we have identified target genes for the tumor suppressor technology to search for p53 target genes in the human genome, protein p53. and have identified several previously described p53 targets in p53 is a sequence-specific DNA-binding protein that regulates addition to numerous potentially novel targets, including the DNA transcription of genes, causing cell-cycle arrest or apoptosis in mismatch repair genes MLH1 and PMS2. Both of these genes were response to DNA damage (15, 16). -

Colon Cancer
Key Antibodies For Colon Cancer Colorectal cancer, also known as bowel cancer, is the 4th most common in the UK. Early Bowel cancer survival rates in In UK are excellent where 95% of men and almost 100% of women will survive stage 1 cancer for 5 years or more after they have been diagnosed. In stage 4 cancer, the 5 year survival rate is only 5% for men and 10% for women, showing the importance of early detection and diagnosis. Menarini Diagnostics is proud to offer our key colon antibodies that aid in the identification of colon cancer. Cancer Research UK (2020), https://www.cancerresearchuk.org/about-cancer/bowel-cancer Key Antibodies for Colon Cancer Product Name Source Clone Catalog Number CDH17 1H3 ACI3111 CDX2 EP25 ACI3144 CDX2 + CDH17 (Double stain) CDX2-88 + EP86 API3135 Cytokeratin 20 (CK20) Ks20.8 CM062A MLH-1 G168-15 ACI3214 MSH2 FE11 CM219 MSH6 BC/44 ACI3215 PMS2 A16-4 CM344 ERCC1 4F9 ACI3147 Key Antibodies for Colon CDH17 CDX2 CDX2 + CDH17 CDH17 is a highly specific marker in colon CDX2 is a sensitive marker for colonic Compared to CDX2 or CK20 alone, the cancer and is a more sensitive marker than carcinoma metastatic to the ovary and combination of CDX2 and CDH17 is highly CDX2 and CK20. It may be helpful for early is more specific than CK20 as it is not sensitive and somewhat specific for colorectal diagnosis of Barrett’s esophagus. CDH17 expressed by serous and endometrioid and stomach adenocarcinoma, especially in staining is positive in the majority of colorectal carcinomas. -

Next-Generation Sequencing Expanded NGS Gene List
Next-Generation Sequencing Expanded NGS Gene List Mutations (DNA) ABI1 BRD4 CRLF2 FOXO4 HOXC11 KLF4 MUC1 PAK3 RHOH TAL2 ABL1 BTG1 DDB2 FSTL3 HOXC13 KLK2 MUTYH PATZ1 RNF213 TBL1XR1 ACKR3 BTK DDIT3 GATA1 HOXD11 LASP1 MYCL (MYCL1) PAX8 RPL10 TCEA1 AKT1 C15orf65 DNM2 GATA2 HOXD13 LMO1 NBN PDE4DIP SEPT5 TCL1A ® AMER1 (FAM123B) CBLC DNMT3A GNA11 HRAS LMO2 NDRG1 PHF6 SEPT6 TERT Tumor Profiling Services from Caris Molecular Intelligence AR CD79B EIF4A2 GPC3 IKBKE MAFB NKX2-1 PHOX2B SFPQ TFE3 ARAF CDH1 ELF4 HEY1 INHBA MAX NONO PIK3CG SLC45A3 TFPT ATP2B3 CDK12 ELN HIST1H3B IRS2 MECOM NOTCH1 PLAG1 SMARCA4 THRAP3 ATRX CDKN2B ERCC1 HIST1H4I JUN MED12 NRAS PMS1 SOCS1 TLX3 BCL11B CDKN2C ETV4 HLF KAT6A (MYST3) MKL1 NUMA1 POU5F1 SOX2 TMPRSS2 BCL2 CEBPA FAM46C HMGN2P46 KAT6B MLLT11 NUTM2B PPP2R1A SPOP UBR5 Next-Generation BCL2L2 CHCHD7 FANCF HNF1A KCNJ5 MN1 OLIG2 PRF1 SRC VHL Sequencing BCOR CNOT3 FEV HOXA11 KDM5C MPL OMD PRKDC SSX1 WAS Multi-platform, solid tumor BCORL1 COL1A1 FOXL2 HOXA13 KDM6A MSN P2RY8 RAD21 STAG2 ZBTB16 Next-Generation Sequencing for BRD3 COX6C FOXO3 HOXA9 KDSR MTCP1 PAFAH1B2 RECQL4 TAL1 ZRSR2 biomarker analysis for therapeutic decision support additional biomarker analysis Mutations and Copy Number Variations (DNA) ABL2 BRCA21 COPB1 ESR1 FUS KIT MYB PER1 RUNX1 TFG ACSL3 BRIP1 CREB1 ETV1 GAS7 KLHL6 MYC PICALM RUNX1T1 TFRC Chemotherapy 4 – ACSL6 BUB1B CREB3L1 ETV5 GATA3 KMT2A (MLL) MYCN PIK3CA SBDS TGFBR2 AFF1 C11orf30 (EMSY) CREB3L2 ETV6 GID4 (C17orf39) KMT2C (MLL3) MYD88 PIK3R1 SDC4 TLX1 Immunotherapy 4 – AFF3 C2orf44 CREBBP EWSR1 -

Lynch Syndrome)
Clinician Management Resource for PMS2 (Lynch syndrome) This overview of clinical management guidelines is based on this patient’s positive test result for a PMS2 gene mutation. Unless otherwise stated, medical management guidelines used here are limited to those issued by the National Comprehensive Cancer Network® (NCCN®)1 in the U.S. Please consult the referenced guideline for complete details and further information. Clinical correlation with the patient’s past medical history, treatments, surgeries and family history may lead to changes in clinical management decisions; therefore, other management recommendations may be considered. Genetic testing results and medical society guidelines help inform medical management decisions but do not constitute formal recommendations. Discussions of medical management decisions and individualized treatment plans should be made in consultation between each patient and his or her healthcare provider, and may change over time. screening/surgical considerations1 age to start frequency Colorectal Cancer 30-35 years old (or 2-5 years prior to the earliest colorectal Colonoscopy** Every 1-2 years cancer in the family, if it is diagnosed before 30 years) Endometrial and Ovarian Cancer Endometrial: encourage prompt response to symptoms (e.g. abnormal uterine bleeding, Individualized Individualized postmenopausal bleeding). Ovarian: educate women on the symptoms associated with ovarian cancer (e.g. pelvic/ Individualized Individualized abdominal pain, bloating, difficulty eating, increased abdominal girth, etc.). Consider option of prophylactic hysterectomy and bilateral salpingo-oophorectomy (BSO) Individualized N/A for women who have completed childbearing Consider screening via endometrial biopsy 30-35 years old Every 1-2 years Transvaginal ultrasound and serum CA-125 may be considered.