Hereditary Cancer Requisition
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Open Full Page
CCR PEDIATRIC ONCOLOGY SERIES CCR Pediatric Oncology Series Recommendations for Childhood Cancer Screening and Surveillance in DNA Repair Disorders Michael F. Walsh1, Vivian Y. Chang2, Wendy K. Kohlmann3, Hamish S. Scott4, Christopher Cunniff5, Franck Bourdeaut6, Jan J. Molenaar7, Christopher C. Porter8, John T. Sandlund9, Sharon E. Plon10, Lisa L. Wang10, and Sharon A. Savage11 Abstract DNA repair syndromes are heterogeneous disorders caused by around the world to discuss and develop cancer surveillance pathogenic variants in genes encoding proteins key in DNA guidelines for children with cancer-prone disorders. Herein, replication and/or the cellular response to DNA damage. The we focus on the more common of the rare DNA repair dis- majority of these syndromes are inherited in an autosomal- orders: ataxia telangiectasia, Bloom syndrome, Fanconi ane- recessive manner, but autosomal-dominant and X-linked reces- mia, dyskeratosis congenita, Nijmegen breakage syndrome, sive disorders also exist. The clinical features of patients with DNA Rothmund–Thomson syndrome, and Xeroderma pigmento- repair syndromes are highly varied and dependent on the under- sum. Dedicated syndrome registries and a combination of lying genetic cause. Notably, all patients have elevated risks of basic science and clinical research have led to important in- syndrome-associated cancers, and many of these cancers present sights into the underlying biology of these disorders. Given the in childhood. Although it is clear that the risk of cancer is rarity of these disorders, it is recommended that centralized increased, there are limited data defining the true incidence of centers of excellence be involved directly or through consulta- cancer and almost no evidence-based approaches to cancer tion in caring for patients with heritable DNA repair syn- surveillance in patients with DNA repair disorders. -

FANCJ Regulates the Stability of FANCD2/FANCI Proteins and Protects Them from Proteasome and Caspase-3 Dependent Degradation
FANCJ regulates the stability of FANCD2/FANCI proteins and protects them from proteasome and caspase-3 dependent degradation Komaraiah Palle, Ph.D. (Kumar) Assistant Professor of Oncologic Sciences Abraham Mitchell Cancer Research Scholar Mitchell Cancer Institute University of South Alabama Outline • Fanconi anemia (FA) pathway • Role of FA pathway in Genome maintenance • FANCJ and FANCD2 functional relationship • FANCJ-mediated DDR in response to Fork-stalling Fanconi Anemia • Rare, inherited blood disorder. • 1:130,000 births Guido Fanconi 1892-1979 • Affects men and women equally. • Affects all racial and ethnic groups – higher incidence in Ashkenazi Jews and Afrikaners Birth Defects Fanconi anemia pathway • FA is a rare chromosome instability syndrome • Autosomal recessive disorder (or X-linked) • Developmental abnormalities • 17 complementation groups identified to date • FA pathway is involved in DNA repair • Increased cancer susceptibility - many patients develop AML - in adults solid tumors Fanconi Anemia is an aplastic anemia FA patients are prone to multiple types of solid tumors • Increased incidence and earlier onset cancers: oral cavity, GI and genital and reproductive tract head and neck breast esophagus skin liver brain Why? FA is a DNA repair disorder • FA caused by mutations in 17 genes: FANCA FANCF FANCM FANCB FANCG/XRCC9 FANCN/PALB2 FANCC FANCI RAD51C/FANCO FANCD1/BRCA2 FANCJ SLX4/FANCP FANCD2 FANCL ERCC2/XPF/FANCQ FANCE BRCA1/FANCS • FA genes function in DNA repair processes • FA patient cells are highly sensitive -

DNA Damage Response and Cancer Therapeutics Through the Lens of the Fanconi Anemia DNA Repair Pathway Sonali Bhattacharjee* and Saikat Nandi*
Bhattacharjee and Nandi Cell Communication and Signaling (2017) 15:41 DOI 10.1186/s12964-017-0195-9 REVIEW Open Access DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway Sonali Bhattacharjee* and Saikat Nandi* Abstract Fanconi Anemia (FA) is a rare, inherited genomic instability disorder, caused by mutations in genes involved in the repair of interstrand DNA crosslinks (ICLs). The FA signaling network contains a unique nuclear protein complex that mediates the monoubiquitylation of the FANCD2 and FANCI heterodimer, and coordinates activities of the downstream DNA repair pathway including nucleotide excision repair, translesion synthesis, and homologous recombination. FA proteins act at different steps of ICL repair in sensing, recognition and processing of DNA lesions. The multi-protein network is tightly regulated by complex mechanisms, such as ubiquitination, phosphorylation, and degradation signals that are critical for the maintenance of genome integrity and suppressing tumorigenesis. Here, we discuss recent advances in our understanding of how the FA proteins participate in ICL repair and regulation of the FA signaling network that assures the safeguard of the genome. We further discuss the potential application of designing small molecule inhibitors that inhibit the FA pathway and are synthetic lethal with DNA repair enzymes that can be used for cancer therapeutics. Keywords: DNA repair, Fanconi Anemia (FA) signaling network, DNA damage response, Cancer therapeutics, Synthetic lethality, Combination Therapy Genomic instability, Interstrand crosslink (ICL), Homologous recombination, Translesion synthesis Background Additional clinical features included microcephaly, vitiligo Fanconi Anemia (FA), a rare genetic cancer-susceptibility and hypoplasia of the testes [8]. After nearly four decades syndrome is a recessive autosomal or X-linked genetic dis- another article reported an accumulation of large number ease [1–3]. -

HEREDITARY CANCER PANELS Part I
Pathology and Laboratory Medicine Clinic Building, K6, Core Lab, E-655 2799 W. Grand Blvd. HEREDITARY CANCER PANELS Detroit, MI 48202 855.916.4DNA (4362) Part I- REQUISITION Required Patient Information Ordering Physician Information Name: _________________________________________________ Gender: M F Name: _____________________________________________________________ MRN: _________________________ DOB: _______MM / _______DD / _______YYYY Address: ___________________________________________________________ ICD10 Code(s): _________________/_________________/_________________ City: _______________________________ State: ________ Zip: __________ ICD-10 Codes are required for billing. When ordering tests for which reimbursement will be sought, order only those tests that are medically necessary for the diagnosis and treatment of the patient. Phone: _________________________ Fax: ___________________________ Billing & Collection Information NPI: _____________________________________ Patient Demographic/Billing/Insurance Form is required to be submitted with this form. Most genetic testing requires insurance prior authorization. Due to high insurance deductibles and member policy benefits, patients may elect to self-pay. Call for more information (855.916.4362) Bill Client or Institution Client Name: ______________________________________________________ Client Code/Number: _____________ Bill Insurance Prior authorization or reference number: __________________________________________ Patient Self-Pay Call for pricing and payment options Toll -

Exploring the Role of Mutations in Fanconi Anemia Genes in Hereditary Cancer Patients
cancers Article Exploring the Role of Mutations in Fanconi Anemia Genes in Hereditary Cancer Patients Jesús del Valle 1,2,3 , Paula Rofes 1,2,3 , José Marcos Moreno-Cabrera 1,2,3 , Adriana López-Dóriga 4,5, Sami Belhadj 1,2,3 , Gardenia Vargas-Parra 1,2,3, Àlex Teulé 1,2,6, Raquel Cuesta 1,2,3, Xavier Muñoz 1,2,3, Olga Campos 1,2,3,Mónica Salinas 1,2, Rafael de Cid 7, Joan Brunet 1,2,3, Sara González 1,2,3, Gabriel Capellá 1,2,3 , Marta Pineda 1,2,3 , Lídia Feliubadaló 1,2,3 and Conxi Lázaro 1,2,3,* 1 Hereditary Cancer Program, Catalan Institute of Oncology, IDIBELL-IGTP-IDIBGI, 08908 Hospitalet de Llobregat, Spain; [email protected] (J.d.V.); [email protected] (P.R.); [email protected] (J.M.M.-C.); [email protected] (S.B.); [email protected] (G.V.-P.); [email protected] (À.T.); [email protected] (R.C.); [email protected] (X.M.); [email protected] (O.C.); [email protected] (M.S.); [email protected] (J.B.); [email protected] (S.G.); [email protected] (G.C.); [email protected] (M.P.); [email protected] (L.F.) 2 Program in Molecular Mechanisms and Experimental Therapy in Oncology (Oncobell), IDIBELL, 08908 Hospitalet de Llobregat, Spain 3 Centro de Investigación Biomédica en Red de Cáncer (CIBERONC), 28029 Madrid, Spain 4 Oncology Data Analytics Program (ODAP), Catalan Institute of Oncology, 08908 Hospitalet de Llobregat, Spain; [email protected] 5 Consortium for Biomedical Research in Epidemiology and Public Health (CIBERESP), 28029 Madrid, Spain 6 Medical Oncology Department, Catalan Institute of Oncology, IDIBELL, 08908 Hospitalet de Llobregat, Spain 7 Genomes for Life—GCAT lab Group, Institut Germans Trias i Pujol (IGTP), 08916 Badalona, Spain; [email protected] * Correspondence: [email protected]; Tel.: +34-93-2607145 Received: 7 February 2020; Accepted: 25 March 2020; Published: 30 March 2020 Abstract: Fanconi anemia (FA) is caused by biallelic mutations in FA genes. -

Mismatch Repair Gene PMS2: Disease-Causing Germline
[CANCER RESEARCH 64, 4721–4727, July 15, 2004] Mismatch Repair Gene PMS2: Disease-Causing Germline Mutations Are Frequent in Patients Whose Tumors Stain Negative for PMS2 Protein, but Paralogous Genes Obscure Mutation Detection and Interpretation Hidewaki Nakagawa,1 Janet C. Lockman,1 Wendy L. Frankel,2 Heather Hampel,1 Kelle Steenblock,3 Lawrence J. Burgart,3 Stephen N. Thibodeau,3 and Albert de la Chapelle1 1Division of Human Cancer Genetics, Comprehensive Cancer Center, and 2Department of Surgical Pathology, The Ohio State University, Columbus, Ohio; and 3Departments of Laboratory Medicine and Pathology, Mayo Clinic and Foundation, Rochester, Minnesota ABSTRACT derived from a family described by Trimbath et al. (3). In two additional cases, children with cancer were heterozygous for germline ␣ The MutL heterodimer formed by mismatch repair (MMR) proteins mutations and even showed widespread microsatellite instability in MLH1 and PMS2 is a major component of the MMR complex, yet normal tissues, but the evidence appeared to support the notion of mutations in the PMS2 gene are rare in the etiology of hereditary non- polyposis colorectal cancer. Evidence from five published cases suggested recessive inheritance (even though a second mutation was not found), that contrary to the Knudson principle, PMS2 mutations cause hereditary in that a parent who had the same mutation had no cancer (4, 5). The nonpolyposis colorectal cancer or Turcot syndrome only when they are fifth patient reported was heterozygous for a PMS2 germline muta- biallelic in the germline or abnormally expressed. As candidates for PMS2 tion, but clinical features were not described (6). mutations, we selected seven patients whose colon tumors stained negative The Knudson two-hit model (7) applies to the MLH1, MSH2, and for PMS2 and positive for MLH1 by immunohistochemistry. -

Epigenetic Regulation of DNA Repair Genes and Implications for Tumor Therapy ⁎ ⁎ Markus Christmann , Bernd Kaina
Mutation Research-Reviews in Mutation Research xxx (xxxx) xxx–xxx Contents lists available at ScienceDirect Mutation Research-Reviews in Mutation Research journal homepage: www.elsevier.com/locate/mutrev Review Epigenetic regulation of DNA repair genes and implications for tumor therapy ⁎ ⁎ Markus Christmann , Bernd Kaina Department of Toxicology, University of Mainz, Obere Zahlbacher Str. 67, D-55131 Mainz, Germany ARTICLE INFO ABSTRACT Keywords: DNA repair represents the first barrier against genotoxic stress causing metabolic changes, inflammation and DNA repair cancer. Besides its role in preventing cancer, DNA repair needs also to be considered during cancer treatment Genotoxic stress with radiation and DNA damaging drugs as it impacts therapy outcome. The DNA repair capacity is mainly Epigenetic silencing governed by the expression level of repair genes. Alterations in the expression of repair genes can occur due to tumor formation mutations in their coding or promoter region, changes in the expression of transcription factors activating or Cancer therapy repressing these genes, and/or epigenetic factors changing histone modifications and CpG promoter methylation MGMT Promoter methylation or demethylation levels. In this review we provide an overview on the epigenetic regulation of DNA repair genes. GADD45 We summarize the mechanisms underlying CpG methylation and demethylation, with de novo methyl- TET transferases and DNA repair involved in gain and loss of CpG methylation, respectively. We discuss the role of p53 components of the DNA damage response, p53, PARP-1 and GADD45a on the regulation of the DNA (cytosine-5)- methyltransferase DNMT1, the key enzyme responsible for gene silencing. We stress the relevance of epigenetic silencing of DNA repair genes for tumor formation and tumor therapy. -

Deficiency in DNA Mismatch Repair of Methylation Damage Is a Major
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.18.388108; this version posted November 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Deficiency in DNA mismatch repair of methylation damage is a major 2 mutational process in cancer 3 1 1 1 1 1,* 4 Hu Fang , Xiaoqiang Zhu , Jieun Oh , Jayne A. Barbour , Jason W. H. Wong 5 6 1School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, The University of 7 Hong Kong, Hong Kong Special Administrative Region 8 9 *Correspondence: [email protected] 10 11 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.18.388108; this version posted November 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Abstract 2 DNA mismatch repair (MMR) is essential for maintaining genome integrity with its 3 deficiency predisposing to cancer1. MMR is well known for its role in the post- 4 replicative repair of mismatched base pairs that escape proofreading by DNA 5 polymerases following cell division2. Yet, cancer genome sequencing has revealed that 6 MMR deficient cancers not only have high mutation burden but also harbour multiple 7 mutational signatures3, suggesting that MMR has pleotropic effects on DNA repair. The 8 mechanisms underlying these mutational signatures have remained unclear despite 9 studies using a range of in vitro4,5 and in vivo6 models of MMR deficiency. -

Involvement of MBD4 Inactivation in Mismatch Repair-Deficient Tumorigenesis
Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Tricarico, R., S. Cortellino, A. Riccio, S. Jagmohan-Changur, H. van der Klift, J. Wijnen, D. Turner, et al. 2015. “Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis.” Oncotarget 6 (40): 42892-42904. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:26318553 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA www.impactjournals.com/oncotarget/ Oncotarget, Vol. 6, No. 40 Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis Rossella Tricarico1, Salvatore Cortellino2, Antonio Riccio3, Shantie Jagmohan- Changur4, Heleen Van der Klift5, Juul Wijnen5, David Turner6, Andrea Ventura7, Valentina Rovella8, Antonio Percesepe9, Emanuela Lucci-Cordisco10, Paolo Radice11, Lucio Bertario11, Monica Pedroni12, Maurizio Ponz de Leon12, Pietro Mancuso1,13, Karthik Devarajan14, Kathy Q. Cai15, Andres J.P. Klein-Szanto15, Giovanni Neri10, Pål Møller16, Alessandra Viel17, Maurizio Genuardi10, Riccardo Fodde4, Alfonso Bellacosa1 1Cancer Epigenetics and Cancer Biology Programs, Fox Chase Cancer Center, Philadelphia, Pennsylvania, United States 2IFOM-FIRC Institute of Molecular Oncology, Milan, -

Predisposition to Hematologic Malignancies in Patients With
LETTERS TO THE EDITOR carcinomas but no internal cancer by the age of 29 years Predisposition to hematologic malignancies in and 9 years, respectively. patients with xeroderma pigmentosum Case XP540BE . This patient had a highly unusual pres - entation of MPAL. She was diagnosed with XP at the age Germline predisposition is a contributing etiology of of 18 months with numerous lentigines on sun-exposed hematologic malignancies, especially in children and skin, when her family emigrated from Morocco to the young adults. Germline predisposition in myeloid neo - USA. The homozygous North African XPC founder muta - plasms was added to the World Health Organization tion was present. 10 She had her first skin cancer at the age 1 2016 classification, and current management recommen - of 8 years, and subsequently developed more than 40 cuta - dations emphasize the importance of screening appropri - neous basal and squamous cell carcinomas, one melanoma 2 ate patients. Rare syndromes of DNA repair defects can in situ , and one ocular surface squamous neoplasm. She 3 lead to myeloid and/or lymphoid neoplasms. Here, we was diagnosed with a multinodular goiter at the age of 9 describe our experience with hematologic neoplasms in years eight months, with several complex nodules leading the defective DNA repair syndrome, xeroderma pigmen - to removal of her thyroid gland. Histopathology showed tosum (XP), including myelodysplastic syndrome (MDS), multinodular adenomatous/papillary hyperplasia. At the secondary acute myeloid leukemia (AML), high-grade age of 19 years, she presented with night sweats, fatigue, lymphoma, and an extremely unusual presentation of and lymphadenopathy. Laboratory studies revealed pancy - mixed phenotype acute leukemia (MPAL) with B, T and topenia with hemoglobin 6.8 g/dL, platelet count myeloid blasts. -
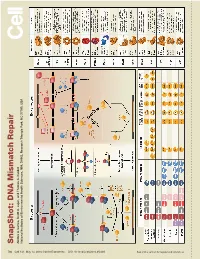
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

DNA Mismatch Repair Proteins MLH1 and PMS2 Can Be Imported to the Nucleus by a Classical Nuclear Import Pathway
Accepted Manuscript DNA mismatch repair proteins MLH1 and PMS2 can be imported to the nucleus by a classical nuclear import pathway Andrea C. de Barros, Agnes A.S. Takeda, Thiago R. Dreyer, Adrian Velazquez- Campoy, Boštjan Kobe, Marcos R.M. Fontes PII: S0300-9084(17)30308-5 DOI: 10.1016/j.biochi.2017.11.013 Reference: BIOCHI 5321 To appear in: Biochimie Received Date: 25 May 2017 Accepted Date: 22 November 2017 Please cite this article as: Keplinger M, Marhofer P, Moriggl B, Zeitlinger M, Muehleder-Matterey S, Marhofer D, Cutaneous innervation of the hand: clinical testing in volunteers shows that variability is far greater than claimed in textbooks, British Journal of Anaesthesia (2017), doi: 10.1016/j.bja.2017.09.008. This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. ACCEPTED MANUSCRIPT ABSTRACT MLH1 and PMS2 proteins form the MutL α heterodimer, which plays a major role in DNA mismatch repair (MMR) in humans. Mutations in MMR-related proteins are associated with cancer, especially with colon cancer. The N-terminal region of MutL α comprises the N-termini of PMS2 and MLH1 and, similarly, the C-terminal region of MutL α is composed by the C-termini of PMS2 and MLH1, and the two are connected by linker region.