Precession Electron Diffraction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Reciprocal Lattice
Appendix A THE RECIPROCAL LATTICE When the translations of a primitive space lattice are denoted by a, b and c, the vector p to any lattice point is given p = ua + vb + we. The definition of the reciprocal lattice is that the translations a*, b* and c*, which define the reciprocal lattice fulfil the following relationships: (A. I) a*.b = b*.c = c*.a = a.b* = b.c* =c. a*= 0 (A.2) It can then be easily shown that: (1) Ic* I = 1/c-spacing of primitive lattice and similarly forb* and a*. (2) a*= (b 1\ c)ja.(b 1\ c) b* = (c 1\ a)jb.(c 1\ a) c* =(a 1\ b)/c-(a 1\ b) These last three relations are often used as a definition of the reciprocal lattice. Two properties of the reciprocal lattice are particularly important: (a) The vector g* defined by g* = ha* + kb* + lc* (where h, k and I are integers to the point hkl in the reciprocal lattice) is normal to the plane of Miller indices (hkl) in the primary lattice. (b) The magnitude Ig* I of this vector is the reciprocal of the spacing of (hkl) in the primary lattice. AI ALLOWED REFLECTIONS The kinematical structure factor for reflections is given by Fhkl = I;J;exp[- 2ni(hu; + kv; + lw)] (A.3) where ui' v; and W; are the coordinates of the atoms and hkl the Miller indices of the reflection g. If there is only one atom at 0, 0, 0 in the unit cell then the structure factor will be independent of hkl since for all values of h, k and I we have Fhkl =f. -

Introduction to Phasing Crystallography ISSN 0907-4449
research papers Acta Crystallographica Section D Biological Introduction to phasing Crystallography ISSN 0907-4449 Garry L. Taylor When collecting X-ray diffraction data from a crystal, we Received 30 August 2009 measure the intensities of the diffracted waves scattered from Accepted 22 February 2010 a series of planes that we can imagine slicing through the Centre for Biomolecular Sciences, University of St Andrews, St Andrews, Fife KY16 9ST, crystal in all directions. From these intensities we derive the Scotland amplitudes of the scattered waves, but in the experiment we lose the phase information; that is, how we offset these waves when we add them together to reconstruct an image of our Correspondence e-mail: [email protected] molecule. This is generally known as the ‘phase problem’. We can only derive the phases from some knowledge of the molecular structure. In small-molecule crystallography, some basic assumptions about atomicity give rise to relationships between the amplitudes from which phase information can be extracted. In protein crystallography, these ab initio methods can only be used in the rare cases in which there are data to at least 1.2 A˚ resolution. For the majority of cases in protein crystallography phases are derived either by using the atomic coordinates of a structurally similar protein (molecular replacement) or by finding the positions of heavy atoms that are intrinsic to the protein or that have been added (methods such as MIR, MIRAS, SIR, SIRAS, MAD, SAD or com- binations of these). The pioneering work of Perutz, Kendrew, Blow, Crick and others developed the methods of isomor- phous replacement: adding electron-dense atoms to the protein without disturbing the protein structure. -

The 4 Reciprocal Lattice
4 The Reciprocal Lattice by Andr~ Authier This electronic edition may be freely copied and redistributed for educational or research purposes only. It may not be sold for profit nor incorporated in any product sold for profit without the express pernfission of The Executive Secretary, International Union of Crystallography, 2 Abbey Square, Chester CIII 211[;, [;K Copyright in this electronic ectition (<.)2001 International l.Jnion of Crystallography Published for the International Union of Crystallography by University College Cardiff Press Cardiff, Wales © 1981 by the International Union of Crystallography. All rights reserved. Published by the University College Cardiff Press for the International Union of Crystallography with the financial assistance of Unesco Contract No. SC/RP 250.271 This pamphlet is one of a series prepared by the Commission on Crystallographic Teaching of the International Union of Crystallography, under the General Editorship of Professor C. A. Taylor. Copies of this pamphlet and other pamphlets in the series may be ordered direct from the University College Cardiff Press, P.O. Box 78, Cardiff CF1 1XL, U.K. ISBN 0 906449 08 I Printed in Wales by University College, Cardiff. Series Preface The long term aim of the Commission on Crystallographic Teaching in establishing this pamphlet programme is to produce a large collection of short statements each dealing with a specific topic at a specific level. The emphasis is on a particular teaching approach and there may well, in time, be pamphlets giving alternative teaching approaches to the same topic. It is not the function of the Commission to decide on the 'best' approach but to make all available so that teachers can make their own selection. -

Appendix F. Glossary
Appendix F. Glossary 2DEG 2-dimensional electron gas A/D Analog to digital AAAR American Association for Aerosol Research ADC Analog-digital converter AEM Analytical electron microscopy AFM Atomic force microscope/microscopy AFOSR Air Force Office of Scientific Research AIST (Japan) Agency of Industrial Science and Technology AIST (Japan, MITI) Agency of Industrial Science and Technology AMLCD Active matrix liquid crystal display AMM Amorphous microporous mixed (oxides) AMO Atomic, molecular, and optical AMR Anisotropic magnetoresistance ARO (U.S.) Army Research Office ARPES Angle-resolved photoelectron spectroscopy ASET (Japan) Association of Super-Advanced Electronics Technologies ASTC Australia Science and Technology Council ATP (Japan) Angstrom Technology Partnership ATP Adenosine triphosphate B Magnetic flux density B/H loop Closed figure showing B (magnetic flux density) compared to H (magnetic field strength) in a magnetizable material—also called hysteresis loop bcc Body-centered cubic BMBF (Germany) Ministry of Education, Science, Research, and Technology (formerly called BMFT) BOD-FF Bond-order-dependent force field BRITE/EURAM Basic Research of Industrial Technologies for Europe, European Research on Advanced Materials program CAD Computer-assisted design CAIBE Chemically assisted ion beam etching CBE Chemical beam epitaxy 327 328 Appendix F. Glossary CBED Convergent beam electron diffraction cermet Ceramic/metal composite CIP Cold isostatic press CMOS Complementary metal-oxide semiconductor CMP Chemical mechanical polishing -
Structure Factors Again
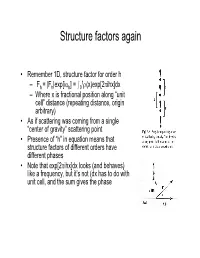
Structure factors again • Remember 1D, structure factor for order h 1 –Fh = |Fh|exp[iαh] = I0 ρ(x)exp[2πihx]dx – Where x is fractional position along “unit cell” distance (repeating distance, origin arbitrary) • As if scattering was coming from a single “center of gravity” scattering point • Presence of “h” in equation means that structure factors of different orders have different phases • Note that exp[2πihx]dx looks (and behaves) like a frequency, but it’s not (dx has to do with unit cell, and the sum gives the phase Back and Forth • Fourier sez – For any function f(x), there is a “transform” of it which is –F(h) = Ûf(x)exp(2pi(hx))dx – Where h is reciprocal of x (1/x) – Structure factors look like that • And it works backward –f(x) = ÛF(h)exp(-2pi(hx))dh – Or, if h comes only in discrete points –f(x) = SF(h)exp(-2pi(hx)) Structure factors, cont'd • Structure factors are a "Fourier transform" - a sum of components • Fourier transforms are reversible – From summing distribution of ρ(x), get hth order of diffraction – From summing hth orders of diffraction, get back ρ(x) = Σ Fh exp[-2πihx] Two dimensional scattering • In Frauenhofer diffraction (1D), we considered scattering from points, along the line • In 2D diffraction, scattering would occur from lines. • Numbering of the lines by where they cut the edges of a unit cell • Atom density in various lines can differ • Reflections now from planes Extension to 3D – Planes defined by extension from 2D case – Unit cells differ • Depends on arrangement of materials in 3D lattice • = "Space -

System Design and Verification of the Precession Electron Diffraction Technique
NORTHWESTERN UNIVERSITY System Design and Verification of the Precession Electron Diffraction Technique A DISSERTATION SUBMITTED TO THE GRADUATE SCHOOL IN PARTIAL FULFILLMENT OF THE REQUIREMENTS for the degree DOCTOR OF PHILOSOPHY Field of Materials Science and Engineering By Christopher Su-Yan Own EVANSTON, ILLINOIS First published on the WWW 01, August 2005 Build 05.12.07. PDF available for download at: http://www.numis.northwestern.edu/Research/Current/precession.shtml c Copyright by Christopher Su-Yan Own 2005 All Rights Reserved ii ABSTRACT System Design and Verification of the Precession Electron Diffraction Technique Christopher Su-Yan Own Bulk structural crystallography is generally a two-part process wherein a rough starting structure model is first derived, then later refined to give an accurate model of the structure. The critical step is the deter- mination of the initial model. As materials problems decrease in length scale, the electron microscope has proven to be a versatile and effective tool for studying many problems. However, study of complex bulk structures by electron diffraction has been hindered by the problem of dynamical diffraction. This phenomenon makes bulk electron diffraction very sensitive to specimen thickness, and expensive equip- ment such as aberration-corrected scanning transmission microscopes or elaborate methodology such as high resolution imaging combined with diffraction and simulation are often required to generate good starting structures. The precession electron diffraction technique (PED), which has the ability to significantly reduce dynamical effects in diffraction patterns, has shown promise as being a “philosopher’s stone” for bulk electron diffraction. However, a comprehensive understanding of its abilities and limitations is necessary before it can be put into widespread use as a standalone technique. -

Chapter 2 X-Ray Diffraction and Reciprocal Lattice
Chapter 2 X-ray diffraction and reciprocal lattice I. Waves 1. A plane wave is described as Ψ(x,t) = A ei(k⋅x-ωt) A is the amplitude, k is the wave vector, and ω=2πf is the angular frequency. 2. The wave is traveling along the k direction with a velocity c given by ω=c|k|. Wavelength along the traveling direction is given by |k|=2π/λ. 3. When a wave interacts with the crystal, the plane wave will be scattered by the atoms in a crystal and each atom will act like a point source (Huygens’ principle). 4. This formulation can be applied to any waves, like electromagnetic waves and crystal vibration waves; this also includes particles like electrons, photons, and neutrons. A particular case is X-ray. For this reason, what we learn in X-ray diffraction can be applied in a similar manner to other cases. II. X-ray diffraction in real space – Bragg’s Law 1. A crystal structure has lattice and a basis. X-ray diffraction is a convolution of two: diffraction by the lattice points and diffraction by the basis. We will consider diffraction by the lattice points first. The basis serves as a modification to the fact that the lattice point is not a perfect point source (because of the basis). 2. If each lattice point acts like a coherent point source, each lattice plane will act like a mirror. θ θ θ d d sin θ (hkl) -1- 2. The diffraction is elastic. In other words, the X-rays have the same frequency (hence wavelength and |k|) before and after the reflection. -

Optical Ptychographic Phase Tomography
University College London Final year project Optical Ptychographic Phase Tomography Supervisors: Author: Prof. Ian Robinson Qiaoen Luo Dr. Fucai Zhang March 20, 2013 Abstract The possibility of combining ptychographic iterative phase retrieval and computerised tomography using optical waves was investigated in this report. The theoretical background and historic developments of ptychographic phase retrieval was reviewed in the first part of the report. A simple review of the principles behind computerised tomography was given with 2D and 3D simulations in the following chapters. The sample used in the experiment is a glass tube with its outer wall glued with glass microspheres. The tube has a diameter of approx- imately 1 mm and the microspheres have a diameter of 30 µm. The experiment demonstrated the successful recovery of features of the sam- ple with limited resolution. The results could be improved in future attempts. In addition, phase unwrapping techniques were compared and evaluated in the report. This technique could retrieve the three dimensional refractive index distribution of an optical component (ideally a cylindrical object) such as an opitcal fibre. As it is relatively an inexpensive and readily available set-up compared to X-ray phase tomography, the technique can have a promising future for application at large scale. Contents List of Figures i 1 Introduction 1 2 Theory 3 2.1 Phase Retrieval . .3 2.1.1 Phase Problem . .3 2.1.2 The Importance of Phase . .5 2.1.3 Phase Retrieval Iterative Algorithms . .7 2.2 Ptychography . .9 2.2.1 Ptychography Principle . .9 2.2.2 Ptychographic Iterative Engine . -

Lecture Notes
Solid State Physics PHYS 40352 by Mike Godfrey Spring 2012 Last changed on May 22, 2017 ii Contents Preface v 1 Crystal structure 1 1.1 Lattice and basis . .1 1.1.1 Unit cells . .2 1.1.2 Crystal symmetry . .3 1.1.3 Two-dimensional lattices . .4 1.1.4 Three-dimensional lattices . .7 1.1.5 Some cubic crystal structures ................................ 10 1.2 X-ray crystallography . 11 1.2.1 Diffraction by a crystal . 11 1.2.2 The reciprocal lattice . 12 1.2.3 Reciprocal lattice vectors and lattice planes . 13 1.2.4 The Bragg construction . 14 1.2.5 Structure factor . 15 1.2.6 Further geometry of diffraction . 17 2 Electrons in crystals 19 2.1 Summary of free-electron theory, etc. 19 2.2 Electrons in a periodic potential . 19 2.2.1 Bloch’s theorem . 19 2.2.2 Brillouin zones . 21 2.2.3 Schrodinger’s¨ equation in k-space . 22 2.2.4 Weak periodic potential: Nearly-free electrons . 23 2.2.5 Metals and insulators . 25 2.2.6 Band overlap in a nearly-free-electron divalent metal . 26 2.2.7 Tight-binding method . 29 2.3 Semiclassical dynamics of Bloch electrons . 32 2.3.1 Electron velocities . 33 2.3.2 Motion in an applied field . 33 2.3.3 Effective mass of an electron . 34 2.4 Free-electron bands and crystal structure . 35 2.4.1 Construction of the reciprocal lattice for FCC . 35 2.4.2 Group IV elements: Jones theory . 36 2.4.3 Binding energy of metals . -

Form and Structure Factors: Modeling and Interactions Jan Skov Pedersen, Department of Chemistry and Inano Center University of Aarhus Denmark SAXS Lab
Form and Structure Factors: Modeling and Interactions Jan Skov Pedersen, Department of Chemistry and iNANO Center University of Aarhus Denmark SAXS lab 1 Outline • Model fitting and least-squares methods • Available form factors ex: sphere, ellipsoid, cylinder, spherical subunits… ex: polymer chain • Monte Carlo integration for form factors of complex structures • Monte Carlo simulations for form factors of polymer models • Concentration effects and structure factors Zimm approach Spherical particles Elongated particles (approximations) Polymers 2 Motivation - not to replace shape reconstruction and crystal-structure based modeling – we use the methods extensively - alternative approaches to reduce the number of degrees of freedom in SAS data structural analysis (might make you aware of the limited information content of your data !!!) - provide polymer-theory based modeling of flexible chains - describe and correct for concentration effects 3 Literature Jan Skov Pedersen, Analysis of Small-Angle Scattering Data from Colloids and Polymer Solutions: Modeling and Least-squares Fitting (1997). Adv. Colloid Interface Sci. , 70 , 171-210. Jan Skov Pedersen Monte Carlo Simulation Techniques Applied in the Analysis of Small-Angle Scattering Data from Colloids and Polymer Systems in Neutrons, X-Rays and Light P. Lindner and Th. Zemb (Editors) 2002 Elsevier Science B.V. p. 381 Jan Skov Pedersen Modelling of Small-Angle Scattering Data from Colloids and Polymer Systems in Neutrons, X-Rays and Light P. Lindner and Th. Zemb (Editors) 2002 Elsevier -

A Review of Transmission Electron Microscopy of Quasicrystals—How Are Atoms Arranged?
crystals Review A Review of Transmission Electron Microscopy of Quasicrystals—How Are Atoms Arranged? Ruitao Li 1, Zhong Li 1, Zhili Dong 2,* and Khiam Aik Khor 1,* 1 School of Mechanical & Aerospace Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore; [email protected] (R.L.); [email protected] (Z.L.) 2 School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore * Correspondence: [email protected] (Z.D.); [email protected] (K.A.K.); Tel.: +65-6790-6727 (Z.D.); +65-6592-1816 (K.A.K.) Academic Editor: Enrique Maciá Barber Received: 30 June 2016; Accepted: 15 August 2016; Published: 26 August 2016 Abstract: Quasicrystals (QCs) possess rotational symmetries forbidden in the conventional crystallography and lack translational symmetries. Their atoms are arranged in an ordered but non-periodic way. Transmission electron microscopy (TEM) was the right tool to discover such exotic materials and has always been a main technique in their studies since then. It provides the morphological and crystallographic information and images of real atomic arrangements of QCs. In this review, we summarized the achievements of the study of QCs using TEM, providing intriguing structural details of QCs unveiled by TEM analyses. The main findings on the symmetry, local atomic arrangement and chemical order of QCs are illustrated. Keywords: quasicrystal; transmission electron microscopy; symmetry; atomic arrangement 1. Introduction The revolutionary discovery of an Al–Mn compound with 5-fold symmetry by Shechtman et al. [1] in 1982 unveiled a new family of materials—quasicrystals (QCs), which exhibit the forbidden rotational symmetries in the conventional crystallography. -

Introduction to Higher Dimensional Description of Quasicrystal Structures
ISQCS, June 23-27, 2019, Sendai, Tohoku University Introduction to higher dimensional description of quasicrystal structures Hiroyuki Takakura Division of Applied Physics, Faculty of Engineering, Hokkaido University ISQCS, June 23-27, 2019, Sendai, Tohoku University Outline • Diffraction symmetries & Space groups of iQCs • Section method • Fibonacci structure • Icosahedral lattices • Simple models of iQCs • Real iQC structures • Cluster based model of iQCs • Summary ISQCS, June 23-27, 2019, Sendai, Tohoku University Crystal Amorphous Their diffraction patterns ISQCS, June 23-27, 2019, Sendai, Tohoku University Diffraction symmetries and space groups of iQCs ISQCS, June 23-27, 2019, Sendai, Tohoku University X-ray transmission Laue patterns of iQC 2-fold 3-fold 5-fold i-Zn-Mg-Ho F-type ISQCS, June 23-27, 2019, Sendai, Tohoku University Electron diffraction pattern of iQC i-AlMn 1 The arrangement of the diffraction spots is not periodic but quasi-periodic. D.Shechtman et al., Phys.Rev.Lett., 53,1951(1984). ISQCS, June 23-27, 2019, Sendai, Tohoku University Symmetry of iQC 2 Point group 31.72º 5 Order : 120 37.38º 2 5 3 20.90º 3 2 2 Asymmetric region: 6 +10 +15 + m + center ISQCS, June 23-27, 2019, Sendai, Tohoku University X-ray diffraction patterns of iQCs P-type i-Zn-Mg-Ho F-type i-Zn-Mg-Ho 2fy 2fy 5f 5f 3f 3f 2fx 2fx Liner plots ISQCS, June 23-27, 2019, Sendai, Tohoku University X-ray diffraction patterns of iQCs P-type i-Zn-Mg-Ho F-type i-Zn-Mg-Ho 2fy 2fy 5f 5f 3f 3f 2fx All even or all odd for 2fx No reflection condition Log plots ISQCS, June 23-27, 2019, Sendai, Tohoku University Vectors used for indexing 6 Any vectors can be used if all the reflections can be indexed correctly.