WO 2016/188962 Al 1 December 2016 (01.12.2016) P O P C T
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of Earthworm Gut-Associated Microorganisms in the Fate of Prions in Soil
THE ROLE OF EARTHWORM GUT-ASSOCIATED MICROORGANISMS IN THE FATE OF PRIONS IN SOIL Von der Fakultät für Lebenswissenschaften der Technischen Universität Carolo-Wilhelmina zu Braunschweig zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigte D i s s e r t a t i o n von Taras Jur’evič Nechitaylo aus Krasnodar, Russland 2 Acknowledgement I would like to thank Prof. Dr. Kenneth N. Timmis for his guidance in the work and help. I thank Peter N. Golyshin for patience and strong support on this way. Many thanks to my other colleagues, which also taught me and made the life in the lab and studies easy: Manuel Ferrer, Alex Neef, Angelika Arnscheidt, Olga Golyshina, Tanja Chernikova, Christoph Gertler, Agnes Waliczek, Britta Scheithauer, Julia Sabirova, Oleg Kotsurbenko, and other wonderful labmates. I am also grateful to Michail Yakimov and Vitor Martins dos Santos for useful discussions and suggestions. I am very obliged to my family: my parents and my brother, my parents on low and of course to my wife, which made all of their best to support me. 3 Summary.....................................................………………………………………………... 5 1. Introduction...........................................................................................................……... 7 Prion diseases: early hypotheses...………...………………..........…......…......……….. 7 The basics of the prion concept………………………………………………….……... 8 Putative prion dissemination pathways………………………………………….……... 10 Earthworms: a putative factor of the dissemination of TSE infectivity in soil?.………. 11 Objectives of the study…………………………………………………………………. 16 2. Materials and Methods.............................…......................................................……….. 17 2.1 Sampling and general experimental design..................................................………. 17 2.2 Fluorescence in situ Hybridization (FISH)………..……………………….………. 18 2.2.1 FISH with soil, intestine, and casts samples…………………………….……... 18 Isolation of cells from environmental samples…………………………….………. -

NCTC) Bacterial Strain Equivalents to American Type Culture Collection (ATCC) Bacterial Strains
This list shows National Collection of Type Cultures (NCTC) bacterial strain equivalents to American Type Culture Collection (ATCC) bacterial strains. NCTC Number CurrentName ATCC Number NCTC 7212 Acetobacter pasteurianus ATCC 23761 NCTC 10138 Acholeplasma axanthum ATCC 25176 NCTC 10171 Acholeplasma equifetale ATCC 29724 NCTC 10128 Acholeplasma granularum ATCC 19168 NCTC 10172 Acholeplasma hippikon ATCC 29725 NCTC 10116 Acholeplasma laidlawii ATCC 23206 NCTC 10134 Acholeplasma modicum ATCC 29102 NCTC 10188 Acholeplasma morum ATCC 33211 NCTC 10150 Acholeplasma oculi ATCC 27350 NCTC 10198 Acholeplasma parvum ATCC 29892 NCTC 8582 Achromobacter denitrificans ATCC 15173 NCTC 10309 Achromobacter metalcaligenes ATCC 17910 NCTC 10807 Achromobacter xylosoxidans subsp. xylosoxidans ATCC 27061 NCTC 10808 Achromobacter xylosoxidans subsp. xylosoxidans ATCC 17062 NCTC 10809 Achromobacter xylosoxidans subsp. xylosoxidans ATCC 27063 NCTC 12156 Acinetobacter baumannii ATCC 19606 NCTC 10303 Acinetobacter baumannii ATCC 17904 NCTC 7844 Acinetobacter calcoaceticus ATCC 15308 NCTC 12983 Acinetobacter calcoaceticus ATCC 23055 NCTC 8102 acinetobacter dna group 13 ATCC 17903 NCTC 10304 Acinetobacter genospecies 13 ATCC 17905 NCTC 10306 Acinetobacter haemolyticus ATCC 17907 NCTC 10305 Acinetobacter haemolyticus subsp haemolyticus ATCC 17906 NCTC 10308 Acinetobacter johnsonii ATCC 17909 NCTC 10307 Acinetobacter junii ATCC 17908 NCTC 5866 Acinetobacter lwoffii ATCC 15309 NCTC 12870 Actinobacillus delphinicola ATCC 700179 NCTC 8529 Actinobacillus equuli ATCC 19392 -

Legionella Shows a Diverse Secondary Metabolism Dependent on a Broad Spectrum Sfp-Type Phosphopantetheinyl Transferase
Legionella shows a diverse secondary metabolism dependent on a broad spectrum Sfp-type phosphopantetheinyl transferase Nicholas J. Tobias1, Tilman Ahrendt1, Ursula Schell2, Melissa Miltenberger1, Hubert Hilbi2,3 and Helge B. Bode1,4 1 Fachbereich Biowissenschaften, Merck Stiftungsprofessur fu¨r Molekulare Biotechnologie, Goethe Universita¨t, Frankfurt am Main, Germany 2 Max von Pettenkofer Institute, Ludwig-Maximilians-Universita¨tMu¨nchen, Munich, Germany 3 Institute of Medical Microbiology, University of Zu¨rich, Zu¨rich, Switzerland 4 Buchmann Institute for Molecular Life Sciences, Goethe Universita¨t, Frankfurt am Main, Germany ABSTRACT Several members of the genus Legionella cause Legionnaires’ disease, a potentially debilitating form of pneumonia. Studies frequently focus on the abundant number of virulence factors present in this genus. However, what is often overlooked is the role of secondary metabolites from Legionella. Following whole genome sequencing, we assembled and annotated the Legionella parisiensis DSM 19216 genome. Together with 14 other members of the Legionella, we performed comparative genomics and analysed the secondary metabolite potential of each strain. We found that Legionella contains a huge variety of biosynthetic gene clusters (BGCs) that are potentially making a significant number of novel natural products with undefined function. Surprisingly, only a single Sfp-like phosphopantetheinyl transferase is found in all Legionella strains analyzed that might be responsible for the activation of all carrier proteins in primary (fatty acid biosynthesis) and secondary metabolism (polyketide and non-ribosomal peptide synthesis). Using conserved active site motifs, we predict Submitted 29 June 2016 some novel compounds that are probably involved in cell-cell communication, Accepted 25 October 2016 Published 24 November 2016 differing to known communication systems. -

Outline of Presentation Legionella Problem in The
2012/2/9 Legionnaire’s Disease: History, Epidemiology; Assessment and Control of Environmental Outline of Presentation Factors leading to Outbreak Conditions Legionaries’ Disease History Occurrence Nature of Disease Route of Infection Presented at the Pathogenesis HKIOEH Professional Development Seminar Diagnosis and treatment Prevention of infection Understanding the ecology of Legionella February 8, 2012 Control Measures Risk assessment / management Joseph K. Kwan An Outbreak investigation The First Legionnaire’s Disease outbreak It occurred during The 1976 American Legionnaires’ annual meeting at a hotel in Philadelphia, Pennsylvania Upon returning home, 221 got sick, 34 died Investigation revealed a responsible bacteria This Bacterium was named (Legionella pneumophila) Records reveal similar outbreaks 1947 & 1967 It all started here in June 1976 Bellevue-Stratford Hotel in Philadelphia, Pennsylvania, USA Since 1976 Outbreaks continue to Legionella Problem in the USA take place all over the world Between 8000 to 18,000 cases reported each year World-wide Occurrence: 10 to 20 % fatality Range of 1 – 21 cases / million ~ 23% are hospital acquired Europe ~ 4.3 cases / million 30- 40% mortality rate HK ~3 cases / million Accuracy depends on efficiency of recognition and reporting!! ~30,000 patients have died from hospital acquired Legionnaires’ disease in the past 25 years No evidence of transmission from person to person All sources are environment related 1 2012/2/9 The situation in HK Legionella Outbreaks -

The Risk to Human Health from Free-Living Amoebae Interaction with Legionella in Drinking and Recycled Water Systems
THE RISK TO HUMAN HEALTH FROM FREE-LIVING AMOEBAE INTERACTION WITH LEGIONELLA IN DRINKING AND RECYCLED WATER SYSTEMS Dissertation submitted by JACQUELINE MARIE THOMAS BACHELOR OF SCIENCE (HONOURS) AND BACHELOR OF ARTS, UNSW In partial fulfillment of the requirements for the award of DOCTOR OF PHILOSOPHY in ENVIRONMENTAL ENGINEERING SCHOOL OF CIVIL AND ENVIRONMENTAL ENGINEERING FACULTY OF ENGINEERING MAY 2012 SUPERVISORS Professor Nicholas Ashbolt Office of Research and Development United States Environmental Protection Agency Cincinnati, Ohio USA and School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Professor Richard Stuetz School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Doctor Torsten Thomas School of Biotechnology and Biomolecular Sciences Faculty of Science The University of New South Wales Sydney, Australia ORIGINALITY STATEMENT '1 hereby declare that this submission is my own work and to the best of my knowledge it contains no materials previously published or written by another person, or substantial proportions of material which have been accepted for the award of any other degree or diploma at UNSW or any other educational institution, except where due acknowledgement is made in the thesis. Any contribution made to the research by others, with whom 1 have worked at UNSW or elsewhere, is explicitly acknowledged in the thesis. I also declare that the intellectual content of this thesis is the product of my own work, except to the extent that assistance from others in the project's design and conception or in style, presentation and linguistic expression is acknowledged.' Signed ~ ............................ -

Isolation and Identification of Free-Living Amoebae from Tap Water in Sivas, Turkey
Hindawi Publishing Corporation BioMed Research International Volume 2013, Article ID 675145, 8 pages http://dx.doi.org/10.1155/2013/675145 Research Article Isolation and Identification of Free-Living Amoebae from Tap Water in Sivas, Turkey Kübra AçJkalJnCoGkun,1 Semra Özçelik,1 Lütfi Tutar,2 Nazif ElaldJ,3 and Yusuf Tutar4,5 1 Department of Parasitology, Faculty of Medicine, Cumhuriyet University, 58140 Sivas, Turkey 2 Department of Biology, Faculty of Science and Letters, Kahramanmaras¸Sutc¨ ¸u¨ Imam˙ University, 46100 Kahramanmaras, Turkey 3 Department of Infectious Diseases, Faculty of Medicine, Cumhuriyet University, 58140 Sivas, Turkey 4 Department of Biochemistry, Faculty of Pharmacology, Cumhuriyet University, 58140 Sivas, Turkey 5 CUTFAM Research Center, Faculty of Medicine, Cumhuriyet University, 58140 Sivas, Turkey Correspondence should be addressed to Yusuf Tutar; [email protected] Received 9 April 2013; Revised 11 June 2013; Accepted 27 June 2013 Academic Editor: Gernot Zissel Copyright © 2013 Kubra¨ Ac¸ıkalın Cos¸kun et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The present work focuses on a local survey of free-living amoebae (FLA) that cause opportunistic and nonopportunistic infections in humans. Determining the prevalence of FLA in water sources can shine a light on the need to prevent FLA related illnesses. A total of 150 samples of tap water were collected from six districts of Sivas province. The samples were filtered and seeded on nonnutrient agar containing Escherichia coli spread. Thirty-three (22%) out of 150 samples were found to be positive for FLA. -

Evaluation of Duopath Legionella Kit for the Rapid Identification Of
Biocontrol Science, 2007, Vol.12, No.4, 155-158 Note Evaluation of DuopathLegionella Kit for the Rapid Identification of Legionella Strains Isolated from Water Samples HIROAKI INOUE1•–, TOMOKO TAKAMA1, YUKIKO AGAWA1, JUNKO ONODERA1, TOMOKI ISHIMA1, KUNIO AGATA1, KEIKO SAITOH2, AND KATSUNORI HURUHATA3 1 Tsukuba Research Laboratories , Aquas Corporation, 4-4 Midorigahara, Tsukuba, lbaraki 300-2646, Research and Investigation Department, Building2 Manegement Education Center, 1-4-28, Mita, Minato, Tokyo 108-0073, School of Environmental Health, Azabu University, 3 1-17-71 Fuchinobe, Sagamihara, Kanagawa 229-8501, Japan Received 20 August, 2007/Accepted 18 October, 2007 Duopath Legionella (Merck KGaA, Darmstadt, Germany) is a rapid and simple immunochromatographic assay kit for the identification of Legionella species. We evaluated the precision of the kit in identifying 100 strains of Legionella and 35 strains of non-Legionella bacteria isolated from cooling tower and bath water samples. Consequently, of all the Legionella strains tested, 99 strains were judged to be Legionella, and only one strain (Legionella busanensis) was judged to be non-Legionella. All of the 35 non-Legionella strains were judged to be non-Legionella. We therefore conclude that Duopath Legionella is a useful method for the rapid identification of Legionella. Key words: Identification/Immunochromatography/Legionella. Legionella species are gram-negative bacteria are required to obtain the final results of the test, be- ubiquitously found in various aquatic environments. If cause the growth of Legionella on the selective agar humans were to inhale aerosolized water from a plates is very slow. Therefore, the development of a source contaminated with Legionella, such as cooling rapid detection and identification procedure for tower or bath water, they could contract a severe Legionella by the culture method is desired. -

Genetic and Functional Studies of the Mip Protein of Legionella
t1.ì. Genetic and Functional Studies of the Mip Protein of Legionella Rodney Mark Ratcliff, BSc (Hons)' MASM Infectious Diseases Laboratories Institute of Medical and Veterinary Science and Department of Microbiology and Immunology UniversitY of Adelaide. Adelaide, South Australia A thesis submitted to the University of Adelaide for the degree of I)octor of Philosophy 15'h March 2000 amended 14th June 2000 Colonies of several Legionella strains on charcoal yeast extract agar (CYE) after 4 days incubation at 37"C in air. Various magnifications show typical ground-glass opalescent appearance. Some pure strains exhibit pleomorphic growth or colour. The top two photographs demonstrate typical red (LH) and blue-white (RH) fluorescence exhibited by some species when illuminated by a Woods (IJV) Lamp. * t Table of Contents .1 Chapter One: Introduction .1 Background .............'. .2 Morphology and TaxonomY J Legionellosis ............. 5 Mode of transmission "..'....'. 7 Environmental habitat 8 Interactions between Legionella and phagocytic hosts 9 Attachment 11 Engulfment and internalisation.'.. 13 Intracellular processing 13 Intracellular replication of legionellae .. " "' " "' 15 Host cell death and bacterial release 18 Virulence (the Genetic factors involved with intracellular multiplication and host cell killing .20 icm/dot system) Legiolysin .25 Msp (Znn* metaloprotease) ...'..... .25 .28 Lipopolysaccharide .29 The association of flagella with disease.. .30 Type IV fimbriae.... .31 Major outer membrane proteins....'.......'. JJ Heat shock proteins'.'. .34 Macrophage infectivity potentiator (Mip) protein Virulenceiraits of Legionella species other than L. pneumophila..........' .39 phylogeny .41 Chapter One (continued): Introduction to bacterial classification and .41 Identificati on of Legionella...'.,..'.. .46 Phylogeny .52 Methods of phylogenetic analysis' .53 Parsimony methods.'.. .55 Distance methods UPGMA cluster analYsis.'.'... -

Electronic Laboratory Reporting Use Case January 2011
ILLINOIS HEALTH INFORMATION EXCHANGE Electronic Laboratory Reporting Use Case Electronic Laboratory Reporting and Health Information Exchange Illinois Health Information Exchange Public Health Work Group January 2011 Electronic Laboratory Reporting Use Case January 2011 Table of Contents 1.0 Executive Summary……………………………………………………………………….3 2.0 Introduction…………………………………………………………………...……………..5 3.0 Scope……………………………………..………………………………………………………5 4.0 Use Case Stakeholders…………………………………………………………….….....6 5.0 Issues and Obstacles……………………………………………………………………...8 6.0 Use Case Pre-Conditions .………………….…………………………………………...8 7.0 Use Case Post-Conditions.……………………………………………………………...9 8.0 Detailed Scenarios/Technical Specifications.………………………………10 9.0 Validation and Certification………………………………………………………...12 Appendix ………………………………………………………………………………………….....13 Page 2 Electronic Laboratory Reporting Use Case January 2011 1.0 Executive Summary This Use Case is a product of the Public Health Work Group (PHWG) of the Illinois Health Information Exchange (HIE) Advisory Committee. The Illinois HIE Advisory Committee was constituted as the diverse public healthcare stakeholder body providing input and recommendations on the creation of the Illinois Health Information Exchange Authority (“the Authority”) as the Illinois vehicle for designing and implementing electronic health information exchange in Illinois. The establishment of the Authority marks the formal transition of the work of the HIE Advisory Committee and the Work Groups into alignment with the provisions of Illinois -

Legionella Pneumophila
Open Research Online The Open University’s repository of research publications and other research outputs Environmental regulation of the growth, physiology and virulence of Legionella pneumophila Thesis How to cite: Mauchline, William Stuart (1996). Environmental regulation of the growth, physiology and virulence of Legionella pneumophila. PhD thesis The Open University. For guidance on citations see FAQs. c 1995 The Author https://creativecommons.org/licenses/by-nc-nd/4.0/ Version: Version of Record Link(s) to article on publisher’s website: http://dx.doi.org/doi:10.21954/ou.ro.0000e0d1 Copyright and Moral Rights for the articles on this site are retained by the individual authors and/or other copyright owners. For more information on Open Research Online’s data policy on reuse of materials please consult the policies page. oro.open.ac.uk ENVIRONMENTAL REGULATION OF THE GROWTH, PHYSIOLOGY AND VIRULENCE OF LEGIONELLA PNEUMOPHILA. WILLIAM STUART MAUCHLINE A thesis submitted in partial fulfilment of the requirements of the Open University for the Degree of Doctor of Philosophy. The research presented in this thesis was undertaken at the Centre for Applied Microbiology and Research, Porton Down, Salisbury. Thames Water Utilities plc was the collaborating establishment. December 1995 APPENDECIES NOT SCANNED AT THE REQUEST OF THE AWARDING UNIVERSITY ABSTRACT Members of the Legionellaceae cause respiratory infections in man; the most severe, pneumonic form is known as Legionnaires' disease. Of the 39 species described to date 16 have been associated with human disease, however the majority of reported cases of legionellosis are caused by Legionella pneumophila serogroup 1. A number of pathogenic bacteria regulate their virulence gene expression in response to environmental stimuli. -

EVALUATION of the P45 MOBILE INTEGRATIVE ELEMENT and ITS ROLE IN
EVALUATION OF THE p45 MOBILE INTEGRATIVE ELEMENT AND ITS ROLE IN Legionella pneumophila VIRULENCE A Dissertation by LANETTE M. CHRISTENSEN Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Chair of Committee, Jeffrey D. Cirillo Committee Members, James Samuel Jon Skare Farida Sohrabji Head of Program, Warren Zimmer May 2018 Major Subject: Medical Sciences Copyright 2018 Lanette Christensen ABSTRACT Legionella pneumophila are aqueous environmental bacilli that live within protozoal species and cause a potentially fatal form of pneumonia called Legionnaires’ disease. Not all L. pneumophila strains have the same capacity to cause disease in humans. The majority of strains that cause clinically relevant Legionnaires’ disease harbor the p45 mobile integrative genomic element. Contribution of the p45 element to L. pneumophila virulence and ability to withstand environmental stress were addressed in this study. The L. pneumophila Philadelphia-1 (Phil-1) mobile integrative element, p45, was transferred into the attenuated strain Lp01 via conjugation, designating p45 an integrative conjugative element (ICE). The resulting trans-conjugate, Lp01+p45, was compared with strains Phil-1 and Lp01 to assess p45 in virulence using a guinea pig model infected via aerosol. The p45 element partially recovered the loss of virulence in Lp01 compared to that of Phil-1 evident in morbidity, mortality, and bacterial burden in the lungs at the time of death. This phenotype was accompanied by enhanced expression of type II interferon in the lungs and spleens 48 hours after infection, independent of bacterial burden. -
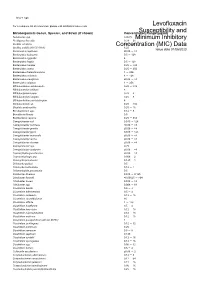
Susceptibility and Resistance Data
toku-e logo For a complete list of references, please visit antibiotics.toku-e.com Levofloxacin Microorganism Genus, Species, and Strain (if shown) Concentration Range (μg/ml)Susceptibility and Aeromonas spp. 0.0625 Minimum Inhibitory Alcaligenes faecalis 0.39 - 25 Bacillus circulans Concentration0.25 - 8 (MIC) Data Bacillus subtilis (ATCC 6051) 6.25 Issue date 01/06/2020 Bacteroides capillosus ≤0.06 - >8 Bacteroides distasonis 0.5 - 128 Bacteroides eggerthii 4 Bacteroides fragilis 0.5 - 128 Bacteroides merdae 0.25 - >32 Bacteroides ovatus 0.25 - 256 Bacteroides thetaiotaomicron 1 - 256 Bacteroides uniformis 4 - 128 Bacteroides ureolyticus ≤0.06 - >8 Bacteroides vulgatus 1 - 256 Bifidobacterium adolescentis 0.25 - >32 Bifidobacterium bifidum 8 Bifidobacterium breve 0.25 - 8 Bifidobacterium longum 0.25 - 8 Bifidobacterium pseudolongum 8 Bifidobacterium sp. 0.25 - >32 Bilophila wadsworthia 0.25 - 16 Brevibacterium spp. 0.12 - 8 Brucella melitensis 0.5 Burkholderia cepacia 0.25 - 512 Campylobacter coli 0.015 - 128 Campylobacter concisus ≤0.06 - >8 Campylobacter gracilis ≤0.06 - >8 Campylobacter jejuni 0.015 - 128 Campylobacter mucosalis ≤0.06 - >8 Campylobacter rectus ≤0.06 - >8 Campylobacter showae ≤0.06 - >8 Campylobacter spp. 0.25 Campylobacter sputorum ≤0.06 - >8 Capnocytophaga ochracea ≤0.06 - >8 Capnocytophaga spp. 0.006 - 2 Chlamydia pneumonia 0.125 - 1 Chlamydia psittaci 0.5 Chlamydia trachomatis 0.12 - 1 Chlamydophila pneumonia 0.5 Citrobacter diversus 0.015 - 0.125 Citrobacter freundii ≤0.00625 - >64 Citrobacter koseri 0.015 -