Contact Potentials, Fermi Level Equilibration and Surface Charging
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Integrated Chemical Microsensor Systems in CMOS Technology by A
microtechnology and mems microtechnology and mems Series Editor: H. Baltes H. Fujita D. Liepmann The series Microtechnology and MEMS comprises text books, monographs, and state-of-the-art reports in the very active field of microsystems and microtech- nology. Written by leading physicists and engineers, the books describe the basic science, device design, and applications. They will appeal to researchers, engineers, and advanced students. Mechanical Microsensors By M. Elwenspoek and R. Wiegerink CMOS Cantilever Sensor Systems Atomic Force Microscopy and Gas Sensing Applications By D. Lange, O. Brand, and H. Baltes Micromachines as Tools for Nanotechnology Editor: H. Fujita Modelling of Microfabrication Systems By R. Nassar and W. Dai Laser Diode Microsystems By H. Zappe Silicon Microchannel Heat Sinks Theories and Phenomena By L. Zhang, K.E. Goodson, and T.W. Kenny Shape Memory Microactuators By M. Kohl Force Sensors for Microelectronic Packaging Applications By J. Schwizer, M. Mayer and O. Brand Integrated Chemical Microsensor Systems in CMOS Technology By A. Hierlemann A. Hierlemann Integrated Chemical Microsensor Systems in CMOS Technology With 125 Figures 123 Professor Dr. Andreas Hierlemann Physical Electronics Laboratory ETH Hoenggerberg, HPT-H 4.2, IQE 8093 Zurich Switzerland Email: [email protected] Series Editors: Professor Dr. H. Baltes ETH Zürich, Physical Electronics Laboratory ETH Hoenggerberg, HPT-H6, 8093 Zürich, Switzerland Professor Dr. Hiroyuki Fujita University of Tokyo, Institute of Industrial Science 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan Professor Dr. Dorian Liepmann University of California, Department of Bioengineering 466 Evans Hall, #1762, Berkeley, CA 94720-1762, USA ISSN 1439-6599 ISBN 3-540-23782-8 Springer Berlin Heidelberg New York LibraryofCongressControlNumber:2004114045 This work is subject to copyright. -

Quantum Mechanics Electromotive Force
Quantum Mechanics_Electromotive force . Electromotive force, also called emf[1] (denoted and measured in volts), is the voltage developed by any source of electrical energy such as a batteryor dynamo.[2] The word "force" in this case is not used to mean mechanical force, measured in newtons, but a potential, or energy per unit of charge, measured involts. In electromagnetic induction, emf can be defined around a closed loop as the electromagnetic workthat would be transferred to a unit of charge if it travels once around that loop.[3] (While the charge travels around the loop, it can simultaneously lose the energy via resistance into thermal energy.) For a time-varying magnetic flux impinging a loop, theElectric potential scalar field is not defined due to circulating electric vector field, but nevertheless an emf does work that can be measured as a virtual electric potential around that loop.[4] In a two-terminal device (such as an electrochemical cell or electromagnetic generator), the emf can be measured as the open-circuit potential difference across the two terminals. The potential difference thus created drives current flow if an external circuit is attached to the source of emf. When current flows, however, the potential difference across the terminals is no longer equal to the emf, but will be smaller because of the voltage drop within the device due to its internal resistance. Devices that can provide emf includeelectrochemical cells, thermoelectric devices, solar cells and photodiodes, electrical generators,transformers, and even Van de Graaff generators.[4][5] In nature, emf is generated whenever magnetic field fluctuations occur through a surface. -

Fundamentals of Electrochemistry
ffirs.qxd 10/29/2005 11:56 AM Page iii FUNDAMENTALS OF ELECTROCHEMISTRY Second Edition V. S. BAGOTSKY A. N. Frumkin Institute of Physical Chemistry and Electrochemistry Russian Academy of Sciences Moscow, Russia Sponsored by THE ELECTROCHEMICAL SOCIETY, INC. Pennington, New Jersey A JOHN WILEY & SONS, INC., PUBLICATION ftoc.qxd 10/29/2005 12:01 PM Page xiv ffirs.qxd 10/29/2005 11:55 AM Page i FUNDAMENTALS OF ELECTROCHEMISTRY ffirs.qxd 10/29/2005 11:55 AM Page ii THE ELECTROCHEMICAL SOCIETY SERIES The Electrochemical Society 65 South Main Street Pennington, NJ 08534-2839 http://www.electrochem.org A complete list of the titles in this series appears at the end of this volume. ffirs.qxd 10/29/2005 11:56 AM Page iii FUNDAMENTALS OF ELECTROCHEMISTRY Second Edition V. S. BAGOTSKY A. N. Frumkin Institute of Physical Chemistry and Electrochemistry Russian Academy of Sciences Moscow, Russia Sponsored by THE ELECTROCHEMICAL SOCIETY, INC. Pennington, New Jersey A JOHN WILEY & SONS, INC., PUBLICATION ffirs.qxd 10/29/2005 11:56 AM Page iv Copyright © 2006 by John Wiley & Sons, Inc. All rights reserved Published by John Wiley & Sons, Inc., Hoboken, New Jersey Published simultaneously in Canada No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning, or otherwise, except as permitted under Section 107 or 108 of the 1976 United States Copyright Act, without either the prior written permission of the Publisher, or authorization through payment of the appropriate per-copy fee to the Copyright Clearance Center, Inc., 222 Rosewood Drive, Danvers, MA 01923, (978) 750-8400, fax (978) 750-4470, or on the web at www.copyright.com. -

Chapter 4: Bonding in Solids and Electronic Properties
Chapter 4: Bonding in Solids and Electronic Properties Free electron theory Consider free electrons in a metal – an electron gas. •regards a metal as a box in which electrons are free to move. •assumes nuclei stay fixed on their lattice sites surrounded by core electrons, while the valence electrons move freely through the solid. •Ignoring the core electrons, one can treat the outer electrons with a quantum mechanical description. •Taking just one electron, the problem is reduced to a particle in a box. Electron is confined to a line of length a. Schrodinger equation 2 2 2 d d 2me E 2 (E V ) 2 2 2me dx dx Electron is not allowed outside the box, so the potential is ∞ outside the box. Energy is quantized, with quantum numbers n. 2 2 2 n2h2 h2 n n n In 1d: E In 3d: E a b c 2 8m a2 b2 c2 8mea e 1 2 2 2 2 Each set of quantum numbers n , n , h n n n a b a b c E 2 2 2 and nc will give rise to an energy level. 8me a b c •In three dimensions, there are multiple combination of energy levels that will give the same energy, whereas in one-dimension n and –n are equal in energy. 2 2 2 2 2 2 na/a nb/b nc/c na /a + nb /b + nc /c 6 6 6 108 The number of states with the 2 2 10 108 same energy is known as the degeneracy. 2 10 2 108 10 2 2 108 When dealing with a crystal with ~1020 atoms, it becomes difficult to work out all the possible combinations. -
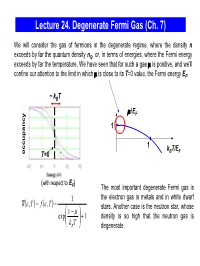
Lecture 24. Degenerate Fermi Gas (Ch
Lecture 24. Degenerate Fermi Gas (Ch. 7) We will consider the gas of fermions in the degenerate regime, where the density n exceeds by far the quantum density nQ, or, in terms of energies, where the Fermi energy exceeds by far the temperature. We have seen that for such a gas μ is positive, and we’ll confine our attention to the limit in which μ is close to its T=0 value, the Fermi energy EF. ~ kBT μ/EF 1 1 kBT/EF occupancy T=0 (with respect to E ) F The most important degenerate Fermi gas is 1 the electron gas in metals and in white dwarf nε()(),, T= f ε T = stars. Another case is the neutron star, whose ε⎛ − μ⎞ exp⎜ ⎟ +1 density is so high that the neutron gas is ⎝kB T⎠ degenerate. Degenerate Fermi Gas in Metals empty states ε We consider the mobile electrons in the conduction EF conduction band which can participate in the charge transport. The band energy is measured from the bottom of the conduction 0 band. When the metal atoms are brought together, valence their outer electrons break away and can move freely band through the solid. In good metals with the concentration ~ 1 electron/ion, the density of electrons in the electron states electron states conduction band n ~ 1 electron per (0.2 nm)3 ~ 1029 in an isolated in metal electrons/m3 . atom The electrons are prevented from escaping from the metal by the net Coulomb attraction to the positive ions; the energy required for an electron to escape (the work function) is typically a few eV. -

Chapter 13 Ideal Fermi
Chapter 13 Ideal Fermi gas The properties of an ideal Fermi gas are strongly determined by the Pauli principle. We shall consider the limit: k T µ,βµ 1, B � � which defines the degenerate Fermi gas. In this limit, the quantum mechanical nature of the system becomes especially important, and the system has little to do with the classical ideal gas. Since this chapter is devoted to fermions, we shall omit in the following the subscript ( ) that we used for the fermionic statistical quantities in the previous chapter. − 13.1 Equation of state Consider a gas ofN non-interacting fermions, e.g., electrons, whose one-particle wave- functionsϕ r(�r) are plane-waves. In this case, a complete set of quantum numbersr is given, for instance, by the three cartesian components of the wave vector �k and thez spin projectionm s of an electron: r (k , k , k , m ). ≡ x y z s Spin-independent Hamiltonians. We will consider only spin independent Hamiltonian operator of the type ˆ 3 H= �k ck† ck + d r V(r)c r†cr , �k � where thefirst and the second terms are respectively the kinetic and th potential energy. The summation over the statesr (whenever it has to be performed) can then be reduced to the summation over states with different wavevectork(p=¯hk): ... (2s + 1) ..., ⇒ r � �k where the summation over the spin quantum numberm s = s, s+1, . , s has been taken into account by the prefactor (2s + 1). − − 159 160 CHAPTER 13. IDEAL FERMI GAS Wavefunctions in a box. We as- sume that the electrons are in a vol- ume defined by a cube with sidesL x, Ly,L z and volumeV=L xLyLz. -

A Fixed Surface Potential Probe with the Swing Capacitive Electrometer
Proc. 2017 Annual Meeting of the Electrostatics of America 1 A Fixed Surface Potential Probe with the Swing Capacitive Electrometer Compared to the Vibrating Kelvin Probe Michael Reznikov Dept. of Integrated Systems Physical Optics Corp., Torrance, CA phone: (1) 310-320-3088 e-mail: [email protected] Abstract— The novel fixed (non-vibrating) Volta potential probe was evaluated in compar- ison with the standard, vibrating (400 Hz) commercial Kelvin probe. While both tested probes demonstrated the similar behavior, there were significant practical advantages of the novel probe. Specifically, the novel probe allows for the elevated distance to the surface and provides more sensitivity to charge under the coating layer. These differences are explained by the analysis of probe circuitry together with the electrostatic approach to the dynamics of the Volta potential measurement. I. INTRODUCTION The capacitive probe method for the measurement of charge induced in the probe elec- trode was proposed by Lord Kelvin (W. Thomson) in 1898 as a capacitive quadrant elec- trometer switching between the immovable probe and discharge shunt while the biasing voltage is applied to the material specimen [1] (see Fig. 1(a)). The using of biasing volt- age and thus the "zero signal" measurement were implemented because the capacitance of the quadrant electrometer is varied during the measurement and thus the measured value depends on the capacitance between the probe electrode and the surface of the specimen. Due to the manual switching and adjustment of biasing voltage, the using of original Kelvin probe was very time-consuming.. In 1932 the vibrating probe method was developed by W.A. -

Thermoelectric Phenomena
Journal of Materials Education Vol. 36 (5-6): 175 - 186 (2014) THERMOELECTRIC PHENOMENA Witold Brostow a, Gregory Granowski a, Nathalie Hnatchuk a, Jeff Sharp b and John B. White a, b a Laboratory of Advanced Polymers & Optimized Materials (LAPOM), Department of Materials Science and Engineering and Department of Physics, University of North Texas, 3940 North Elm Street, Denton, TX 76207, USA; http://www.unt.edu/LAPOM/; [email protected], [email protected], [email protected]; [email protected] b Marlow Industries, Inc., 10451 Vista Park Road, Dallas, TX 75238-1645, USA; Jeffrey.Sharp@ii- vi.com ABSTRACT Potential usefulness of thermoelectric (TE) effects is hard to overestimate—while at the present time the uses of those effects are limited to niche applications. Possibly because of this situation, coverage of TE effects, TE materials and TE devices in instruction in Materials Science and Engineering is largely perfunctory and limited to discussions of thermocouples. We begin a series of review articles to remedy this situation. In the present article we discuss the Seebeck effect, the Peltier effect, and the role of these effects in semiconductor materials and in the electronics industry. The Seebeck effect allows creation of a voltage on the basis of the temperature difference. The twin Peltier effect allows cooling or heating when two materials are connected to an electric current. Potential consequences of these phenomena are staggering. If a dramatic improvement in thermoelectric cooling device efficiency were achieved, the resulting elimination of liquid coolants in refrigerators would stop one contribution to both global warming and destruction of the Earth's ozone layer. -

MOS Capacitors –Theoretical Exercise 3
Semiconductor Device Theory: MOS Capacitors –Theoretical Exercise 3 Dragica Vasileska and Gerhard Klimeck (ASU, Purdue) 1. For an MOS structure, find the charge per unit area in fast surface states ( Qs) as a function of the surface potential ϕs for the surface states density uniformly distributed in energy through- 11 -2 -1 out the forbidden gap with density Dst = 10 cm eV , located at the boundary between the oxide and semiconductor. Assume that the surface states with energies above the neutral level φ0 are acceptor type and that surface states with energies below the neutral level φ0 are donor type. Also assume that all surface states below the Fermi level are filled and that all surface states with energies above the Fermi level are empty (zero-temperature statistics for the sur- face states). The energy gap is Eg=1.12 eV, the neutral level φ0 is 0.3 eV above the valence 10 -3 band edge, the intrinsic carrier concentration is ni=1.5×10 cm , and the semiconductor dop- 15 -3 ing is NA=10 cm . Temperature equals T=300 K. 2. Express the capacitance of an MOS structure per unit area in the presence of uniformly dis- tributed fast surface states (as described in the previous problem) in terms of the oxide ca- pacitance Cox , per unit area. The semiconductor capacitance equals to Cs=-dQs/d ϕs (where Qs is the charge in the semiconductor and ϕs is the surface potential), and the surface states ca- pacitance is defined as Css =-dQst /d ϕs, where Qst is the charge in fast surface states per unit area. -

Sixfold Fermion Near the Fermi Level in Cubic Ptbi2 Abstract Contents
SciPost Phys. 10, 004 (2021) Sixfold fermion near the Fermi level in cubic PtBi2 Setti Thirupathaiah1, 2, Yevhen Kushnirenko1, Klaus Koepernik1, 3, Boy Roman Piening1, Bernd Büchner1, 3, Saicharan Aswartham1, Jeroen van den Brink1, 3, Sergey Borisenko1 and Ion Cosma Fulga1, 3? 1 IFW Dresden, Helmholtzstr. 20, 01069 Dresden, Germany 2 Condensed Matter Physics and Material Sciences Department, S. N. Bose National Centre for Basic Sciences, Kolkata, West Bengal-700106, India 3 Würzburg-Dresden Cluster of Excellence ct.qmat, Germany ? [email protected] Abstract We show that the cubic compound PtBi2 is a topological semimetal hosting a sixfold band touching point in close proximity to the Fermi level. Using angle-resolved photoemission spectroscopy, we map the band structure of the system, which is in good agreement with results from density functional theory. Further, by employing a low energy effective Hamiltonian valid close to the crossing point, we study the effect of a magnetic field on the sixfold fermion. The latter splits into a total of twenty Weyl cones for a Zeeman field oriented in the diagonal, (111) direction. Our results mark cubic PtBi2 as an ideal candidate to study the transport properties of gapless topological systems beyond Dirac and Weyl semimetals. Copyright S. Thirupathaiah et al. Received 17-06-2020 This work is licensed under the Creative Commons Accepted 03-12-2020 Check for Attribution 4.0 International License. Published 11-01-2021 updates Published by the SciPost Foundation. doi:10.21468/SciPostPhys.10.1.004 Contents 1 Introduction1 2 Band structure mapping2 2.1 Theory 2 2.2 Experiment4 3 Low energy model7 4 Conclusion 9 A Model parameters from ab-initio data 10 References 11 1 SciPost Phys. -

Electroplating, Electrochemistry and Electronics
NASF SURFACE TECHNOLOGY WHITE PAPERS 80 (10), 1-44 (July 2016) The 15th William Blum Lecture Presented at the 61st AES Annual Convention in Chicago, Illinois June 17, 1974 Electroplating, Electrochemistry and Electronics by George Dubpernell M&T Chemicals Ferndale, Michigan Recipient of the 1973 William Blum AES Scientific Achievement Award Page 1 NASF SURFACE TECHNOLOGY WHITE PAPERS 80 (10), 1-44 (July 2016) Contents 1. Historical 3 2. The Periodic Chart 4 3. The consumption of metals in electroplating 5 4. The double standard of electrode potentials - pH and reference electrodes 8 5. On the nature of electrode potentials and hydrogen overvoltage 14 6. Experimental 15 7. Discussion 21 8. Hydrogen overvoltage in electroplating 27 9. Contact potential - Volta potential - Electrostatic surface potential 30 10. The so-called hydrogen electrode 33 11. The Nernst theory of the electromotive activity of ions 34 12. Electrophysiology 35 13. Relationships to electronics 36 14. References 38 15. About the author 43 Page 2 NASF SURFACE TECHNOLOGY WHITE PAPERS 80 (10), 1-44 (July 2016) The 15th William Blum Lecture Presented at the 61st AES Annual Convention in Chicago, Illinois June 17, 1974 Electroplating, Electrochemistry and Electronics by George Dubpernell M&T Chemicals Ferndale, Michigan Recipient of the 1973 William Blum AES Scientific Achievement Award Editor’s Note: Originally published as Plating, 62 (4), 327-334 (1975), Plating, 62 (5), 436-442 (1975) and Plating, 62 (6), 573- 580 (1975), this article is a re-publication of the 15th William Blum Lecture, presented at the 61st AES Annual Convention in Chicago, Illinois on June 17, 1974. -

Interfacial Potentials in Ion Solvation
Interfacial Potentials in Ion Solvation A dissertation submitted to the Graduate School of the University of Cincinnati in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Physics of the McMicken College of Arts and Sciences by Carrie Conor Doyle B.S. in Physics, Rutgers, the State University of New Jersey, 2013 June 2020 supervised by Dr. Thomas L. Beck Committee Co-Chair: Dr. Carlos Bolech, Physics Committee Member: Dr. Rohana Wiedjewardhana, Physics Committee Member: Dr. Leigh Smith, Physics Abstract Solvation science is an integral part of many fields across physics, chemistry, and biology. Liquids, interfaces, and the ions that populate them are responsible for many poorly understood natural phenomena such as ion specific effects. Establishing a single-ion solvation free energy thermodynamic scale is a necessary component to unraveling ion-specific effects. This task is made difficult by the experimental immeasurability of quantities such as the interfacial potential between two media, which sets the scale. Computer simulations provide a necessary bridge between experimental and theoretical results. However, computer models are limited by the accuracy-efficiency dilemma, and results are misinterpreted when the underlying physics is overlooked. Classical molecular dynamic techniques, while efficient, lack transferability. Quantum-based ab initio techniques are accurate and transferable, but their inefficiency limits the accessible simulation size and time. This thesis seeks to determine the physical origin of the interfacial potential at the liquid-vapor interface using classical models. Additionally, I assess the ability of Neural Network Potential (NNP) simulation methods to produce electrostatic properties of bulk liquids and interfaces.