ELECTRONIC STRUCTURE of the SEMIMETALS Bi and Sb 1571
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Quantum Mechanics Electromotive Force
Quantum Mechanics_Electromotive force . Electromotive force, also called emf[1] (denoted and measured in volts), is the voltage developed by any source of electrical energy such as a batteryor dynamo.[2] The word "force" in this case is not used to mean mechanical force, measured in newtons, but a potential, or energy per unit of charge, measured involts. In electromagnetic induction, emf can be defined around a closed loop as the electromagnetic workthat would be transferred to a unit of charge if it travels once around that loop.[3] (While the charge travels around the loop, it can simultaneously lose the energy via resistance into thermal energy.) For a time-varying magnetic flux impinging a loop, theElectric potential scalar field is not defined due to circulating electric vector field, but nevertheless an emf does work that can be measured as a virtual electric potential around that loop.[4] In a two-terminal device (such as an electrochemical cell or electromagnetic generator), the emf can be measured as the open-circuit potential difference across the two terminals. The potential difference thus created drives current flow if an external circuit is attached to the source of emf. When current flows, however, the potential difference across the terminals is no longer equal to the emf, but will be smaller because of the voltage drop within the device due to its internal resistance. Devices that can provide emf includeelectrochemical cells, thermoelectric devices, solar cells and photodiodes, electrical generators,transformers, and even Van de Graaff generators.[4][5] In nature, emf is generated whenever magnetic field fluctuations occur through a surface. -

Novel Electronic Properties of Monoclinic
www.nature.com/scientificreports OPEN Novel electronic properties of monoclinic M P4 (M = Cr, Mo, W) compounds with or without topological nodal line Muhammad Rizwan Khan1,2, Kun Bu1,2, Jun‑Shuai Chai1,2 & Jian‑Tao Wang1,2,3* Transition metal phosphides hold novel metallic, semimetallic, and semiconducting behaviors. Here we report by ab initio calculations a systematical study on the structural and electronic properties of 6 MP4 (M = Cr, Mo, W) phosphides in monoclinic C2/c (C 2h ) symmetry. Their dynamical stabilities have been confrmed by phonon modes calculations. Detailed analysis of the electronic band structures and density of states reveal that CrP4 is a semiconductor with an indirect band gap of 0.47 eV in association with the p orbital of P atoms, while MoP4 is a Dirac semimetal with an isolated nodal point at the Ŵ point and WP4 is a topological nodal line semimetal with a closed nodal ring inside the frst Brillouin zone relative to the d orbital of Mo and W atoms, respectively. Comparison of the phosphides with group VB, VIB and VIIB transition metals shows a trend of change from metallic to semiconducting behavior from VB-MP4 to VIIB-MP4 compounds. These results provide a systematical understandings on the distinct electronic properties of these compounds. Transition metal phosphides (TMPs) have been attracted considerable research interest due to their structural and compositional diversity that results in a broad range of novel electronic, magnetic and catalytic properties1–4. Tis family consists of large number of materials, having distinct crystallographic structures and morpholo- gies because of choices of diferent TMs and phosphorus atoms 5. -

Hear the Sound of Weyl Fermions
PHYSICAL REVIEW X 9, 021053 (2019) Featured in Physics Hear the Sound of Weyl Fermions Zhida Song1,2 and Xi Dai1,* 1Department of Physics, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong 2Department of Physics, Princeton University, Princeton, New Jersey 08544, USA (Received 4 February 2019; revised manuscript received 10 April 2019; published 17 June 2019) Quasiparticles and collective modes are two fundamental aspects that characterize quantum matter in addition to its ground-state features. For example, the low-energy physics for Fermi-liquid phase in He-III is featured not only by fermionic quasiparticles near the chemical potential but also by fruitful collective modes in the long-wave limit, including several different sound waves that can propagate through it under different circumstances. On the other hand, it is very difficult for sound waves to be carried by electron liquid in ordinary metals due to the fact that long-range Coulomb interaction among electrons will generate a plasmon gap for ordinary electron density oscillation and thus prohibits the propagation of sound waves through it. In the present paper, we propose a unique type of acoustic collective mode in Weyl semimetals under magnetic field called chiral zero sound. Chiral zero sound can be stabilized under the so-called “chiral limit,” where the intravalley scattering time is much shorter than the intervalley one and propagates only along an external magnetic field for Weyl semimetals with multiple pairs of Weyl points. The sound velocity of chiral zero sound is proportional to the field strength in the weak field limit, whereas it oscillates dramatically in the strong field limit, generating an entirely new mechanism for quantum oscillations through the dynamics of neutral bosonic excitation, which may manifest itself in the thermal conductivity measurements under magnetic field. -

Chapter 4: Bonding in Solids and Electronic Properties
Chapter 4: Bonding in Solids and Electronic Properties Free electron theory Consider free electrons in a metal – an electron gas. •regards a metal as a box in which electrons are free to move. •assumes nuclei stay fixed on their lattice sites surrounded by core electrons, while the valence electrons move freely through the solid. •Ignoring the core electrons, one can treat the outer electrons with a quantum mechanical description. •Taking just one electron, the problem is reduced to a particle in a box. Electron is confined to a line of length a. Schrodinger equation 2 2 2 d d 2me E 2 (E V ) 2 2 2me dx dx Electron is not allowed outside the box, so the potential is ∞ outside the box. Energy is quantized, with quantum numbers n. 2 2 2 n2h2 h2 n n n In 1d: E In 3d: E a b c 2 8m a2 b2 c2 8mea e 1 2 2 2 2 Each set of quantum numbers n , n , h n n n a b a b c E 2 2 2 and nc will give rise to an energy level. 8me a b c •In three dimensions, there are multiple combination of energy levels that will give the same energy, whereas in one-dimension n and –n are equal in energy. 2 2 2 2 2 2 na/a nb/b nc/c na /a + nb /b + nc /c 6 6 6 108 The number of states with the 2 2 10 108 same energy is known as the degeneracy. 2 10 2 108 10 2 2 108 When dealing with a crystal with ~1020 atoms, it becomes difficult to work out all the possible combinations. -
Lecture 24. Degenerate Fermi Gas (Ch
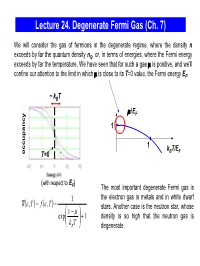
Lecture 24. Degenerate Fermi Gas (Ch. 7) We will consider the gas of fermions in the degenerate regime, where the density n exceeds by far the quantum density nQ, or, in terms of energies, where the Fermi energy exceeds by far the temperature. We have seen that for such a gas μ is positive, and we’ll confine our attention to the limit in which μ is close to its T=0 value, the Fermi energy EF. ~ kBT μ/EF 1 1 kBT/EF occupancy T=0 (with respect to E ) F The most important degenerate Fermi gas is 1 the electron gas in metals and in white dwarf nε()(),, T= f ε T = stars. Another case is the neutron star, whose ε⎛ − μ⎞ exp⎜ ⎟ +1 density is so high that the neutron gas is ⎝kB T⎠ degenerate. Degenerate Fermi Gas in Metals empty states ε We consider the mobile electrons in the conduction EF conduction band which can participate in the charge transport. The band energy is measured from the bottom of the conduction 0 band. When the metal atoms are brought together, valence their outer electrons break away and can move freely band through the solid. In good metals with the concentration ~ 1 electron/ion, the density of electrons in the electron states electron states conduction band n ~ 1 electron per (0.2 nm)3 ~ 1029 in an isolated in metal electrons/m3 . atom The electrons are prevented from escaping from the metal by the net Coulomb attraction to the positive ions; the energy required for an electron to escape (the work function) is typically a few eV. -

Chapter 13 Ideal Fermi
Chapter 13 Ideal Fermi gas The properties of an ideal Fermi gas are strongly determined by the Pauli principle. We shall consider the limit: k T µ,βµ 1, B � � which defines the degenerate Fermi gas. In this limit, the quantum mechanical nature of the system becomes especially important, and the system has little to do with the classical ideal gas. Since this chapter is devoted to fermions, we shall omit in the following the subscript ( ) that we used for the fermionic statistical quantities in the previous chapter. − 13.1 Equation of state Consider a gas ofN non-interacting fermions, e.g., electrons, whose one-particle wave- functionsϕ r(�r) are plane-waves. In this case, a complete set of quantum numbersr is given, for instance, by the three cartesian components of the wave vector �k and thez spin projectionm s of an electron: r (k , k , k , m ). ≡ x y z s Spin-independent Hamiltonians. We will consider only spin independent Hamiltonian operator of the type ˆ 3 H= �k ck† ck + d r V(r)c r†cr , �k � where thefirst and the second terms are respectively the kinetic and th potential energy. The summation over the statesr (whenever it has to be performed) can then be reduced to the summation over states with different wavevectork(p=¯hk): ... (2s + 1) ..., ⇒ r � �k where the summation over the spin quantum numberm s = s, s+1, . , s has been taken into account by the prefactor (2s + 1). − − 159 160 CHAPTER 13. IDEAL FERMI GAS Wavefunctions in a box. We as- sume that the electrons are in a vol- ume defined by a cube with sidesL x, Ly,L z and volumeV=L xLyLz. -

Density Functional Theory
Density Functional Theory Fundamentals Video V.i Density Functional Theory: New Developments Donald G. Truhlar Department of Chemistry, University of Minnesota Support: AFOSR, NSF, EMSL Why is electronic structure theory important? Most of the information we want to know about chemistry is in the electron density and electronic energy. dipole moment, Born-Oppenheimer charge distribution, approximation 1927 ... potential energy surface molecular geometry barrier heights bond energies spectra How do we calculate the electronic structure? Example: electronic structure of benzene (42 electrons) Erwin Schrödinger 1925 — wave function theory All the information is contained in the wave function, an antisymmetric function of 126 coordinates and 42 electronic spin components. Theoretical Musings! ● Ψ is complicated. ● Difficult to interpret. ● Can we simplify things? 1/2 ● Ψ has strange units: (prob. density) , ● Can we not use a physical observable? ● What particular physical observable is useful? ● Physical observable that allows us to construct the Hamiltonian a priori. How do we calculate the electronic structure? Example: electronic structure of benzene (42 electrons) Erwin Schrödinger 1925 — wave function theory All the information is contained in the wave function, an antisymmetric function of 126 coordinates and 42 electronic spin components. Pierre Hohenberg and Walter Kohn 1964 — density functional theory All the information is contained in the density, a simple function of 3 coordinates. Electronic structure (continued) Erwin Schrödinger -

Nonsaturating Large Magnetoresistance in Semimetals
This document is downloaded from DR‑NTU (https://dr.ntu.edu.sg) Nanyang Technological University, Singapore. Nonsaturating large magnetoresistance in semimetals Leahy, Ian A.; Lin, Yu‑Ping; Siegfried, Peter E.; Treglia, Andrew C.; Song, Justin Chien Wen; Nandkishore, Rahul M.; Lee, Minhyea 2018 Leahy, I. A., Lin, Y.‑P., Siegfried, P. E., Treglia, A. C., Song, J. C. W., Nandkishore, R. M., & Lee, M. (2018). Nonsaturating large magnetoresistance in semimeta. Proceedings of the National Academy of Sciences of the United States of America, 115 (42), 10570‑10575. doi:10.1073/pnas.1808747115 https://hdl.handle.net/10356/137998 https://doi.org/10.1073/pnas.1808747115 © 2018 The Author(s). All rights reserved. This paper was published by National Academy of Sciences in Proceedings of the National Academy of Sciences of the United States of America and is made available with permission of The Author(s). Downloaded on 01 Oct 2021 03:16:44 SGT Non-saturating large magnetoresistance in semimetals Ian A. Leahy,1 Yu-Ping Lin,1 Peter E. Siegfried,1 Andrew C. Treglia,1 Justin C. W. Song,2 Rahul M. Nandkishore,1, 3 and Minhyea Lee1, 4, ∗ 1Department of Physics, University of Colorado, Boulder, CO 80309, USA 2Division of Physics and Applied Physics, Nanyang Technological University, Singapore 637371 3Center for Theory of Quantum Matter, University of Colorado, Boulder, CO 80309, USA 4Center for Experiments on Quantum Materials, University of Colorado, Boulder, CO 80309, USA (Dated: September 6, 2018) The rapidly expanding class of quantum materials known as topological semimetals (TSM) dis- play unique transport properties, including a striking dependence of resistivity on applied magnetic field, that are of great interest for both scientific and technological reasons. -

It's Been a Weyl Coming
CONDENSED MATTER It’s been a Weyl coming Condensed-matter physics brings us quasiparticles that behave as massless fermions. B. Andrei Bernevig “Mathematizing may well be a creative activity of man, like language or music”1 — so said Hermann Weyl, the German physicist whose penchant for mathematical elegance prompted his prediction that a new particle would arise when the fermionic mass in the Dirac equation vanished2. Such a particle could carry charge but, unlike all known fermions, would be massless. During the course of his career, Weyl actually fell out of love with his prediction, largely because it implied the breaking of a particular symmetry, known as parity, which at the time was thought to be obeyed. More to the point, no such particle was observed during his lifetime. After his death, the Weyl fermion was proposed to describe neutrinos, which are now known to have mass. For some time, it seemed that the Weyl fermion was destined to be just an abstract concept from another beautiful mind. That was until the Weyl fermion entered the realm of condensed-matter physics. For several years this field has been considered fertile ground for finding the Weyl fermion. Now, three papers in Nature Physics3–5 have cemented earlier findings6,7 to confirm the predictions8,9 of Weyl physics in a family of nonmagnetic materials with broken inversion symmetry. In condensed-matter physics, specifically in solid-state band structures, Weyl fermions appear when two electronic bands cross. The crossing point is called a Weyl node, away from which the bands disperse linearly in the lattice momentum, giving rise to a special kind of semimetal. -

“Band Structure” for Electronic Structure Description
www.fhi-berlin.mpg.de Fundamentals of heterogeneous catalysis The very basics of “band structure” for electronic structure description R. Schlögl www.fhi-berlin.mpg.de 1 Disclaimer www.fhi-berlin.mpg.de • This lecture is a qualitative introduction into the concept of electronic structure descriptions different from “chemical bonds”. • No mathematics, unfortunately not rigorous. • For real understanding read texts on solid state physics. • Hellwege, Marfunin, Ibach+Lüth • The intention is to give an impression about concepts of electronic structure theory of solids. 2 Electronic structure www.fhi-berlin.mpg.de • For chemists well described by Lewis formalism. • “chemical bonds”. • Theorists derive chemical bonding from interaction of atoms with all their electrons. • Quantum chemistry with fundamentally different approaches: – DFT (density functional theory), see introduction in this series – Wavefunction-based (many variants). • All solve the Schrödinger equation. • Arrive at description of energetics and geometry of atomic arrangements; “molecules” or unit cells. 3 Catalysis and energy structure www.fhi-berlin.mpg.de • The surface of a solid is traditionally not described by electronic structure theory: • No periodicity with the bulk: cluster or slab models. • Bulk structure infinite periodically and defect-free. • Surface electronic structure theory independent field of research with similar basics. • Never assume to explain reactivity with bulk electronic structure: accuracy, model assumptions, termination issue. • But: electronic -

Electronic Properties of Type-II Weyl Semimetal Wte2. a Review Perspective
Electronic properties of type-II Weyl semimetal WTe2. A review perspective. P. K. Das1, D. Di Sante2, F. Cilento3, C. Bigi4, D. Kopic5, D. Soranzio5, A. Sterzi3, J. A. Krieger6,7,8, I. Vobornik9, J. Fujii9, T. Okuda10, V. N. Strocov6, M. B. H. Breese1,11, F. Parmigiani3,5, G. Rossi4,9, S. Picozzi12, R. Thomale2, G. Sangiovanni2, R. J. Cava13, and G. Panaccione9,* 1Singapore Synchrotron Light Source, National University of Singapore, 5 Research Link, 117603, Singapore 2Institut für Theoretische Physik und Astrophysik, Universität Würzburg, Am Hubland Campus Süd, Würzburg 97074, Germany 3Elettra - Sincrotrone Trieste S.C.p.A., Strada Statale 14, km 163.5, Trieste 34149, Italy 4Dipartimento di Fisica, Universitá di Milano, Via Celoria 16, I-20133 Milano, Italy 5Universitá degli Studi di Trieste - Via A. Valerio 2, Trieste 34127, Italy 6Paul Scherrer Institute, Swiss Light Source, CH-5232 Villigen, Switzerland 7Laboratory for Muon Spin Spectroscopy, Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland 8Laboratorium für Festkörperphysik, ETH Zürich, CH-8093 Zürich, Switzerland 9Istituto Officina dei Materiali (IOM)-CNR, Laboratorio TASC, in Area Science Park, S.S.14, Km 163.5, I- 34149 Trieste, Italy 10Hiroshima Synchrotron Radiation Center (HSRC), Hiroshima University, 2-313 Kagamiyama, Higashi- Hiroshima 739-0046, Japan. 11Department of Physics, National University of Singapore, 117576, Singapore 12Consiglio Nazionale delle Ricerche (CNR-SPIN), c/o Univ. Chieti-Pescara "G. D'Annunzio", 66100 Chieti, Italy 13Department of Chemistry, Princeton University, Princeton, New Jersey 08544, USA * Corresponding author: [email protected] Currently, there is a flurry of research interest on materials with an unconventional electronic structure, and we have already seen significant progress in their understanding and engineering towards real-life applications. -

Simple Molecular Orbital Theory Chapter 5
Simple Molecular Orbital Theory Chapter 5 Wednesday, October 7, 2015 Using Symmetry: Molecular Orbitals One approach to understanding the electronic structure of molecules is called Molecular Orbital Theory. • MO theory assumes that the valence electrons of the atoms within a molecule become the valence electrons of the entire molecule. • Molecular orbitals are constructed by taking linear combinations of the valence orbitals of atoms within the molecule. For example, consider H2: 1s + 1s + • Symmetry will allow us to treat more complex molecules by helping us to determine which AOs combine to make MOs LCAO MO Theory MO Math for Diatomic Molecules 1 2 A ------ B Each MO may be written as an LCAO: cc11 2 2 Since the probability density is given by the square of the wavefunction: probability of finding the ditto atom B overlap term, important electron close to atom A between the atoms MO Math for Diatomic Molecules 1 1 S The individual AOs are normalized: 100% probability of finding electron somewhere for each free atom MO Math for Homonuclear Diatomic Molecules For two identical AOs on identical atoms, the electrons are equally shared, so: 22 cc11 2 2 cc12 In other words: cc12 So we have two MOs from the two AOs: c ,1() 1 2 c ,1() 1 2 2 2 After normalization (setting d 1 and _ d 1 ): 1 1 () () [2(1 S )]1/2 12 [2(1 S )]1/2 12 where S is the overlap integral: 01 S LCAO MO Energy Diagram for H2 H2 molecule: two 1s atomic orbitals combine to make one bonding and one antibonding molecular orbital.