Selection and Environmental Adaptation Along a Path to Speciation in the Tibetan Frog Nanorana Parkeri
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Geodetic Model of the 2017 Mw 6.5 Mainling Earthquake Inferred from GPS and Insar Data
remote sensing Letter Geodetic Model of the 2017 Mw 6.5 Mainling Earthquake Inferred from GPS and InSAR Data Huizi Jian, Lifeng Wang *, Weijun Gan, Keliang Zhang, Yanchuan Li , Shiming Liang, Yunhua Liu, Wenyu Gong and Xinzhong Yin State Key Laboratory of Earthquake Dynamics, Institute of Geology, China Earthquake Administration, Beijing 100029, China; [email protected] (H.J.); [email protected] (W.G.); [email protected] (K.Z.); [email protected] (Y.L.); [email protected] (S.L.); [email protected] (Y.L.); [email protected] (W.G.); [email protected] (X.Y.) * Correspondence: [email protected]; Tel.: +86-010-6200-9427 Received: 2 November 2019; Accepted: 3 December 2019; Published: 9 December 2019 Abstract: On 17 November 2017, a Mw 6.5 earthquake occurred in Mainling County, Nyingchi City, China. The epicenter was located in the Namche Barwa region of the eastern Himalayan syntaxis. Here, we have derived coseismic deformation from Global Positioning System (GPS) data and ascending Sentinel-1A Synthetic Aperture Radar (SAR) data. Based on a joint inversion of the two datasets, we obtained the coseismic slip distribution along a curved, northeast trending, and high-angle (dip angle of 75◦) thrust fault. Our results show that the seismic moment release was 7.49 1018 N m, corresponding to a moment magnitude of Mw 6.55. The maximum slip was 1.03 m × · and the main rupture zone extended to a 12 km depth. The earthquake may have been related to the release of strain accumulated during the subduction of the Indian plate beneath the Eurasian continent. -

Battle Against Poverty Being Won in Tibet
6 | Tuesday, September 1, 2020 HONG KONG EDITION | CHINA DAILY CHINA Poverty alleviation Battle against poverty being won in Tibet Major investments in infrastructure and new homes improve life for villagers. Palden Nyima reports from Lhasa. ccess to fresh water used to be a major concern for Tibetan villager Migmar. She had to take a Kyilung Tibet 40-minuteA round trip on a tractor Namling every two days to haul water home Saga in a container across rough terrain. Shigatse Taking showers and doing laundry Layak were luxuries for the community leader and her fellow villagers in CHINA DAILY Saga county in Southwest China’s Tibet autonomous region. mother could get subsidies and sup- Fast forward three years, and port when giving birth in a hospital. Layak village, 180 kilometers from I did not know it could be safer for the county seat in the southwest- both mother and child,” Samdrub ern part of Tibet, now has taps that Tsering said. provide potable water at the “top of The township center also used to the world”. be inaccessible for many villagers. “Our village had no proper roads While the nearest household lives or safe drinking water before 2016. about 10 km away, some families But now, all the families have were 200 km from town, with no access to tap water and the village telecommunication networks avail- is connected by paved roads,” said able. Road conditions were terrible, Migmar, 49, who is the village he said. leader. Thanks to the government’s pov- The roads and pipelines have erty alleviation measures, liveli- helped lay the groundwork for a hoods have improved tremendously significant improvement in the over the years, Samdrub Tsering villagers’ lives, with Layak one of said. -

Report on Domestic Animal Genetic Resources in China
Country Report for the Preparation of the First Report on the State of the World’s Animal Genetic Resources Report on Domestic Animal Genetic Resources in China June 2003 Beijing CONTENTS Executive Summary Biological diversity is the basis for the existence and development of human society and has aroused the increasing great attention of international society. In June 1992, more than 150 countries including China had jointly signed the "Pact of Biological Diversity". Domestic animal genetic resources are an important component of biological diversity, precious resources formed through long-term evolution, and also the closest and most direct part of relation with human beings. Therefore, in order to realize a sustainable, stable and high-efficient animal production, it is of great significance to meet even higher demand for animal and poultry product varieties and quality by human society, strengthen conservation, and effective, rational and sustainable utilization of animal and poultry genetic resources. The "Report on Domestic Animal Genetic Resources in China" (hereinafter referred to as the "Report") was compiled in accordance with the requirements of the "World Status of Animal Genetic Resource " compiled by the FAO. The Ministry of Agriculture" (MOA) has attached great importance to the compilation of the Report, organized nearly 20 experts from administrative, technical extension, research institutes and universities to participate in the compilation team. In 1999, the first meeting of the compilation staff members had been held in the National Animal Husbandry and Veterinary Service, discussed on the compilation outline and division of labor in the Report compilation, and smoothly fulfilled the tasks to each of the compilers. -

News China March. 13.Cdr
VOL. XXV No. 3 March 2013 Rs. 10.00 The first session of the 12th National People’s Congress (NPC) opens at the Great Hall of the People in Beijing, capital of China on March 5, 2013. (Xinhua/Pang Xinglei) Chinese Ambassador to India Mr. Wei Wei meets Indian Chinese Vice Foreign Minister Cheng Guoping , on behalf Foreign Minister Salman Khurshid in New Delhi on of State Councilor Dai Bingguo, attends the dialogue on February 25, 2013. During the meeting the two sides Afghanistan issue held in Moscow,together with Russian exchange views on high-level interactions between the two Security Council Secretary Nikolai Patrushev and Indian countries, economic and trade cooperation and issues of National Security Advisor Shivshankar Menon on February common concern. 20, 2013. Chinese Ambassador to India Mr.Wei Wei and other VIP Chinese Ambassador to India Mr. Wei Wei and Indian guests are having a group picture with actors at the 2013 Minister of Culture Smt. Chandresh Kumari Katoch enjoy Happy Spring Festival organized by the Chinese Embassy “China in the Spring Festival” exhibition at the 2013 Happy and FICCI in New Delhi on February 25,2013. Artists from Spring Festival. The exhibition introduces cultures, Jilin Province, China and Punjab Pradesh, India are warmly customs and traditions of Chinese Spring Festival. welcomed by the audience. Chinese Ambassador to India Mr. Wei Wei(third from left) Chinese Ambassador to India Mr. Wei Wei visits the participates in the “Happy New Year “ party organized by Chinese Visa Application Service Centre based in the Chinese Language Department of Jawaharlal Nehru Southern Delhi on March 6, 2013. -
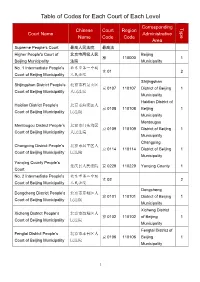
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

The Biggest, the Grandest, Luxurious & the Most Epic
WELCOME The Biggest, The Grandest, Luxurious & The Most Epic Bus Journey’s in The World Introduction An all-encompassing journey of magnificent menus, outstanding scenery and incredible cultural heritage, this itinerary brings you face-to-face with Europe’s & Asia’s finest attractions. Celebrate Catalonia with a small-group pintxo (tapas) tasting, enjoy a tasting of robust Tuscan reds and ascend to the top of Europe on Mount Stanserhorn to learn about regional fauna and flora from a Swiss Ranger. You will meet passionate locals along the way who will share insider knowledge and enrich your experience. See all the 'stans (well, most of them) on this comprehensive Bus tour through Central Asia. Explore natural landscapes like Kaindy Lake's sunken forest and witness the hustle and bustle of capital cities and their bazaars, cathedrals, and historical sites. Along the way, you'll Eat like the locals and Sleep in Yurts / Hotels to get even closer to this underexplored destination. Start in Volgograd and end in Moscow! With the Historical tour Remarkable Russia, you have a Epic Road tour taking you through Volgograd, Russia and Moscow, Remarkable Russia includes accommodation in a hotel as well as an expert guide, meals, transport and more. Treat yourself with the epic Himalayan panorama before enjoying a roller- coaster ride to vibrant Kathmandu. The thrilling Tibet Nepal tours are expertly crafted to fulfill your ultimate fantasy to delve into two of the most fascinating Himalayan Kingdoms across the Mighty Himalayas. we promise you quality one-stop service for the entire journey, with safety guaranteed. -

February 2016
INTERNATIONAL U.S.-Russia Relations 2016 Reg. ss-973 February www.southasia.com.pk INSIDE INDIA SRI LANKA AFGHANISTAN NEIGHBOR Local Focus Sad Saga No Child’s Play The Deal and After ISIS Marches Recent incidentsEast of violence in Afghanistan, Pakistan, Bangladesh and Indonesia indicate that the Islamic State (IS) is moving eastwards from Syria and Iraq. Afghanistan Afg. 50 India Rs. 65 Philippines P 75 Australia A$ 6 Japan ¥ 500 Saudi Arabia SR 15 Bangladesh Taka 65 Korea Won 3000 Singapore S$ 8 Bhutan NU 45 Malaysia RM 6 Sri Lanka Rs. 100 Brazil BRL 20 Maldives Rf 45 Thailand B 100 Canada C$ 6 Myanmar MMK10 Turkey Lira. 2 China RMB 30 Nepal NcRs. 75 UAE AED 10 France Fr 30 New Zealand NZ$ 7 UK £ 3 Hong Kong HK$ 30 Pakistan Rs. 150 USA $ 5 Ad 2 Contents 12 Eastward Bound The IS looks for new territories to conquer. India Sri Lanka 26Local Focus ‘Make In India’ is still Sad Saga groping in the dark. The unwanted dividends of 32 prosperity. Nepal Big Brother Politics Nepal should get better 36 treatment from India. The Maldives Indian Ocean Turmoil 38 The pains of growth. 30 Bangladesh Rising Religious Extremism A surge of Islamic fundamentalism. 4 SOUTHASIA • FEBRUARY 2016 REGULAR FEATURES Editor’s Mail 8 On Record 9 Briefs 10 COVER STORY Eastward Bound 12 IS and Saarc 16 Changing Directions 20 Expanding Territory 22 REGION Pakistan Honour Among Thieves 24 India Local Focus 26 Afghanistan Shattered Country 28 42 Bangladesh International Rising Religious Extremism 30 Hot and Cold Sri Lanka Indications for world peace. -
Tibet Insight, 1-15 June 2018
TIBET INSIGHT, 1-15 JUNE 2018 Page 1 of 21 TAR NEWS TAR Party Secretary visits Kindergarten June 01, 2018 On International Children’s Day on May 31, TAR Party Secretary Wu Yingjie visited a Bilingual kindergarten in Chengguan district of Lhasa. Posters highlighting the contrast between “old and new Tibet” were pasted in the corridors of the school. TAR Party Secretary Wu Yingjie advised the children to understand the trust and loyalty of the Party and to remember Chinese President Xi Jinping's message of safeguarding the motherland's unity. Training Course for Lhasa’s Budget Performance June 05, 2018 A training class for enhancing efficiency in Lhasa’s budget performance began in Lhasa on June 4. Well-known experts were invited, including a Professor from Beijing’s Information Science and Technology University who gave a lecture on financial performance and management. The Training was especially to strengthen the budgetary performance of the Central Party’s special funds. All departments have been mandated to achieve an overall budget performance target of over 300,000 Yuan by end of the fiscal year 2019. The Lhasa Municipal Government has also established a mechanism to assess nine key livelihood policies and major projects in Lhasa. Tibet University Takes Measures to Promote Teachers' Teaching Skills June 05, 2018 Since March 2018, the Tibet University has launched a large number of military training activities for teachers. To oversee the progress of this training, a group comprising “excellent teachers with high moral character, excellent professional skills, and strong educational and teaching abilities” was constituted. According to the arrangement, Tibet University has organized departments to further study the State Council's "Opinions on Comprehensively Deepening the Reform of the Teaching Staff in the New Era", "National Standards for Undergraduate Teaching Quality", "Undergraduate Professional Certification Standards" and "Tibetan College Teacher's Classroom Basic Requirements" etc. -

China - Provisions of Administration on Border Trade of Small Amount and Foreign Economic and Technical Cooperation of Border Regions, 1996
China - Provisions of Administration on Border Trade of Small Amount and Foreign Economic and Technical Cooperation of Border Regions, 1996 MOFTEC Copyright © 1996 MOFTEC ii Contents Contents Article 16 5 Article 17 5 Chapter 1 - General Provisions 2 Article 1 2 Chapter 3 - Foreign Economic and Technical Coop- eration in Border Regions 6 Article 2 2 Article 18 6 Article 3 2 Article 19 6 Chapter 2 - Border Trade of Small Amount 3 Article 20 6 Article 21 6 Article 4 3 Article 22 7 Article 5 3 Article 23 7 Article 6 3 Article 24 7 Article 7 3 Article 25 7 Article 8 3 Article 26 7 Article 9 4 Article 10 4 Chapter 4 - Supplementary Provisions 9 Article 11 4 Article 27 9 Article 12 4 Article 28 9 Article 13 5 Article 29 9 Article 14 5 Article 30 9 Article 15 5 SiSU Metadata, document information 11 iii Contents 1 Provisions of Administration on Border Trade of Small Amount and Foreign Economic and Technical Cooperation of Border Regions (Promulgated by the Ministry of Foreign Trade Economic Cooperation and the Customs General Administration on March 29, 1996) 1 China - Provisions of Administration on Border Trade of Small Amount and Foreign Economic and Technical Cooperation of Border Regions, 1996 2 Chapter 1 - General Provisions 3 Article 1 4 With a view to strengthening and standardizing the administra- tion on border trade of small amount and foreign economic and technical cooperation of border regions, preserving the normal operating order for border trade of small amount and techni- cal cooperation of border regions, and promoting the healthy and steady development of border trade, the present provisions are formulated according to the Circular of the State Council on Circular of the State Council on Certain Questions of Border Trade. -

© 生物多样性biodiversity Science
胡一鸣, 梁健超, 金崑, 丁志锋, 周智鑫, 胡慧建, 蒋志刚. 喜马拉雅山哺乳动物物种多样性垂直分布格局. 生物多样性, 2018, 26 (2): 191–201. http://www.biodiversity-science.net/CN/10.17520/biods.2017324 附录 1 哺乳动物物种数据获取的主要方法 Appendix 1 Methods for acquiring data of mammal species 1.1 野外调查时间与样线布设情况 1.1 Survey area, date of survey and sampling effort 调查区域 调查日期 样线数量 样线总长度 Survey area Date of survey Number of transects Total length of the transects (km) 吉隆县吉隆沟 Gyirong Valley, 2012.05.10-2012.10.13, 46 110.0 Gyirong county 2013.07.09-2013.09.05, 2017.06.20-2017.06.26. 聂拉木县樟木沟 Zhangmu Valley, 2013.07.11-2013.07.20, 22 89.8 Nyalam County 2017.06.16-2017.06.23. 定日县绒线沟 Rongxian Valley, 2013.09.12-2013.09.16. 11 56.7 Tingri County 定日县嘎玛沟 Gama Valley, Tingri 2013.05.15-2013.05.23. 1 62.4 County 定结县陈塘沟 Chentang Valley, 2013.05.15-2013.05.23. 10 91.3 Dinggyê County 亚东县亚东沟 Yadong Valley, 2013.05.16-2013.05.24, 22 91.7 Yadong County 2017.06.10-2017.06.16. 洛扎县多布沟 Duobu Valley, 2013.09.29-2013.10.04. 10 51.3 Lhozhag County 错那县勒布沟 Lubu Valley, Cona 2013.09.28-2013.10.05, 30 58.6 County 2017.06.03-2017.06.09. 错那县浪坡沟 Langpo Valley, Cona 2013.09.26-2013.09.27, 5 29.3 County 2015.06.06-2015.06.07. 隆子县玉麦沟 Yumai Valley, 2013.09.23-2013.09.14, 3 15.0 Lhünzê County 2015.06.04-2015.06.05. 隆子县扎日沟 Zhari Valley, Lhünzê 2013.09.20-2013.09.24, 12 64.8 County 2015.05.30-2015.06.04. -

PLA Joint Exercises in Tibet: Implications for India
FEBRUARY 2020 PLA Joint Exercises in Tibet: Implications for India RAJESWARI PILLAI RAJAGOPALAN PULKIT MOHAN PLA Joint Exercises in Tibet: Implications for India RAJESWARI PILLAI RAJAGOPALAN PULKIT MOHAN ABOUT THE AUTHORS Dr Rajeswari Pillai Rajagopalan is a Distinguished Fellow and Head of Space and Nuclear Policy Initiative at ORF. She is also the senior Asia defence writer for The Diplomat. She is the author of four books: Nuclear Security in India (2015), Clashing Titans: Military Strategy and Insecurity among Asian Great Powers (2012), The Dragon’s Fire: Chinese Military Strategy and Its Implications for Asia (2009), and Uncertain Eagle: US Military Strategy in Asia (2009). Pulkit Mohan is a Junior Fellow at ORF’s Space and Nuclear Policy Initiative. Before joining ORF, Pulkit was an Editorial Assistant with a leading development journal. She obtained her Master’s degree in International Relations from the London School of Economics. ISBN: 978-93-89622-66-9 © 2020 Observer Research Foundation. All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means without permission in writing from ORF. PLA Joint Exercises in Tibet: Implications for India ABSTRACT Since 2015, China has been undertaking a thorough restructuring of its military, under the leadership of President Xi Jinping. This has involved significant changes in the operational structure of the People's Liberation Army (PLA), with the modernisation and optimisation of the military to bring about better jointness and overall efficiency. This paper examines the PLA reforms as they pertain to training and jointness in the Tibet Autonomous Region (TAR) and the Sino-Indian border areas. -

1 Tibet Insight, October-December 2020
TIBET INSIGHT, OCTOBER-DECEMBER 2020 1 TAR NEWS Tibet Insight News: October 2020 Head of TAR UFWD inspects Zezhol Monastery in Chamdo October 30, 2020 On October 28, Danke, Standing Committee Member of TAR Party Committee, Minister of TAR United Front Work Department (UFWD) and Vice-Chairman of TAR Political Consultative Conference (PPCC) inspected Zezhol Monastery, an important and one of the oldest monasteries of the Yundrung Bon sect, in Chamdo. The inspection was to evaluate the monastery’s understanding and implementation of the ‘spirit’ of the Seventh Work Forum on Tibet, investigate monastic management and ascertain the “progress” of patriotic re-education activities especially the “four standards to become exemplary monks and nuns”. At a meeting during his visit, Danke shared that a blueprint of how to build a ‘united, prosperous, harmonious and socialist new Tibet’ was drafted after the Seventh Work Forum on Tibet and the TAR Party Committee’s eighth plenary session. He said the primary political task of religious institutions and representatives, at present and in the future, should be to study in-depth and comprehensively of the spirit of the Tibet Work Forum, Xi Jinping’s “strategic exposition” of governing and stabilising Tibet’s borders, and “sinicization of Tibetan Buddhism”. Briefly, the Head of TAR UFWD stressed the following: 1. The need to resolutely study, implement and carry forward the spirit of all the seven work forums on Tibet, unify political positions, unswervingly follow the path of socialism with Chinese characteristics, safeguard national unity and strengthen all propaganda campaigns; 2. The need to reciprocate the love to the “core”, meaning to Xi Jinping and his “special care and love” for Tibet, thereby improving political and ideological consciousness, 3.