Genome Assembly, Annotation and Comparative Analysis of the Cattail
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

(BCF), Translocation Factor (TF) and Metal Enrichment Factor (MEF) Abilities of Aquatic Macrophyte Species Exposed to Metal Contaminated Wastewater
ISSN(Online): 2319-8753 ISSN (Print): 2347-6710 International Journal of Innovative Research in Science, Engineering and Technology (A High Impact Factor, Monthly, Peer Reviewed Journal) Visit: www.ijirset.com Vol. 8, Issue 1, January 2019 Evaluation of Bioaccumulation Factor (BAF), Bioconcentration Factor (BCF), Translocation Factor (TF) and Metal Enrichment Factor (MEF) Abilities of Aquatic Macrophyte Species Exposed to Metal Contaminated Wastewater S. S. Shingadgaon1, B.L. Chavan2 Research Scholar, Department of Environmental Science, School of Earth Sciences, Solapur University, Solapur, MS, India1 Former Professor and Head, Department of Environmental Science, Solapur University Solapur and presently working at Department of Environmental Science, Dr.Babasaheb Ambedkar Marathwada University, Aurangabad, MS, India 2 ABSTRACT: Wastewaters receiving aquatic bodies are quiet complex in terms of pollutants, the transport and interactions with heavy metals. This complexity is primarily due to high variability of pollutants, contaminants and related parameters. The macrophytes are plausible bio-indicators of the pollution load and level of metals within the aquatic systems than the wastewater or sediment analyses. The potential ability of aquatic macrophytes in natural water bodies receiving municipal sewage from Solapur city was assessed. Data from the studies on macrophytes exposed to a mixed test bath of metals and examined to know their potentialities to accumulate heavy metals for judging their suitability for phytoremediation technology -
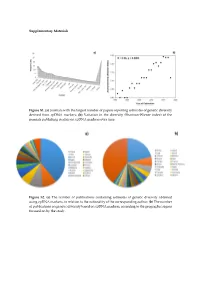
(A) Journals with the Largest Number of Papers Reporting Estimates Of
Supplementary Materials Figure S1. (a) Journals with the largest number of papers reporting estimates of genetic diversity derived from cpDNA markers; (b) Variation in the diversity (Shannon-Wiener index) of the journals publishing studies on cpDNA markers over time. Figure S2. (a) The number of publications containing estimates of genetic diversity obtained using cpDNA markers, in relation to the nationality of the corresponding author; (b) The number of publications on genetic diversity based on cpDNA markers, according to the geographic region focused on by the study. Figure S3. Classification of the angiosperm species investigated in the papers that analyzed genetic diversity using cpDNA markers: (a) Life mode; (b) Habitat specialization; (c) Geographic distribution; (d) Reproductive cycle; (e) Type of flower, and (f) Type of pollinator. Table S1. Plant species identified in the publications containing estimates of genetic diversity obtained from the use of cpDNA sequences as molecular markers. Group Family Species Algae Gigartinaceae Mazzaella laminarioides Angiospermae Typhaceae Typha laxmannii Angiospermae Typhaceae Typha orientalis Angiospermae Typhaceae Typha angustifolia Angiospermae Typhaceae Typha latifolia Angiospermae Araliaceae Eleutherococcus sessiliflowerus Angiospermae Polygonaceae Atraphaxis bracteata Angiospermae Plumbaginaceae Armeria pungens Angiospermae Aristolochiaceae Aristolochia kaempferi Angiospermae Polygonaceae Atraphaxis compacta Angiospermae Apocynaceae Lagochilus macrodontus Angiospermae Polygonaceae Atraphaxis -

Proceedings of the Workshop on the Creation of Channels and Ponds Within Cattail Marshes on the Bay of Quinte, and a Conceptual Plan
PROCEEDINGS OF THE WORKSHOP ON THE CREATION OF CHANNELS AND PONDS WITHIN CATTAIL MARSHES ON THE BAY OF QUINTE, AND A CONCEPTUAL PLAN. PREPARED BY ANDY SMITH BAY OF QUINTE REMEDIAL ACTION PLAN JANUARY, 1995 PREFACE On August 17 and 18, 1994 a workshop was held to bring together scientists and members of , , the Bay of Quinte Implementation Advisory Committee (formally the Public Advisory Committee) to discuss enhancing Quinte wetlands by dredging channels and ponds in dense cattail stands. The goals for the workshop were to review the impacts of this technique, discuss its advantages and disadvantages, and design a new channel/pond system. This report is a summary of the workshop and a conceptual plan for a project based on recommendations from the workshop. If implemented this experimental/demonstration project will be studied to determine the effectiveness of creating open water areas within dense cattail stands for providing habitat for variety of species. TABLE OF CONTENTS 1.0 Workshop Introduction ...................................... 1 2.0 Summary of Workshop Presentations and Discussions:~,' ................. 3 2.1 A Literature Review of the Impacts to Wildlife of Channel Creation Through Monotypic Cattail Stands as Proposed at the Bay of Quinte Area of Concern 3 2.2 Studies Conducted on Wetlands in The Quinte Area, 1994 ........... 4 2.2.1 Review of Some Recent Wetland Enhancement Projects in the Quinte Area and Creation techniques. 4 2.2.2 Fisheries Assessment of Some Wetland Enhancement Projects in the Quinte Area . 5 2.2.3 List of Plants and Animals Observed During the Tour of Sawguin Creek Marsh, August 17, 1994 .......................... -

Studies of the Germination and Growth of Cattail in Relation to Marsh Management John William Bedish Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1964 Studies of the germination and growth of cattail in relation to marsh management John William Bedish Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Terrestrial and Aquatic Ecology Commons Recommended Citation Bedish, John William, "Studies of the germination and growth of cattail in relation to marsh management" (1964). Retrospective Theses and Dissertations. 16857. https://lib.dr.iastate.edu/rtd/16857 This Thesis is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. STUDIES OF THE GERMINATION AND GROWTH OF CATTAIL IN RELATION TO K~SH MANAGEMENT by John William Bedish A Thesis Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of MASTER OF SCIENCE Major Subject: Wildlife Management Signatures have been redacted for privacy Iowa State University Of Science and Technology Ames, Iowa 1964 ii TABLE OF CONTENTS Page INTRODUCTION 1 LITERATURE REVIEW 3 Importance of Cattail to Marsh Animals 3 Relation of Water to Presence of Cattail 5 Seed Germination and Viability 8 METHODS OF STUDY 14 Greenhouse Studies 14 Field Studies 23 RESULTS 29 Greenhouse Studies 29 Field Studies 46 DISCUSSION 65 Effects of Moisture on Cattail 65 Management Recommendations 69 SUMMARY 76 LITERATURE CITED 79 ACKNOWLEDGEMENTS 84 1 INTRODUCTION The total area of wetlands available to waterfowl has been greatly reduced during the past century due mainly to drainage for agricultural purposes. -

Hybridization Dynamics of Invasive Cattail (Typhaceae) Stands at Pierce Cedar Creek Institute: a Molecular Analysis
HYBRIDIZATION DYNAMICS OF INVASIVE CATTAIL (TYPHACEAE) STANDS AT PIERCE CEDAR CREEK INSTITUTE: A MOLECULAR ANALYSIS Alex Graeff, Kelsey Huisman, and Dr. Pamela J. Laureto Department of Biological Sciences Grand Rapids Community College 143 Bostwick NE Grand Rapids, Michigan 49503 ABSTRACT Three cattail taxa are recognized in Michigan USA: native Typha latifolia (broad-leaf cattail), the invasive Typha angustifolia (narrow-leaf cattail), and the hybrid of these two species Typha × glauca. Typha angustifolia and T. × glauca are of special interest because of their ability to aggressively spread and out-compete the native cattail T. latifolia. Typha × glauca has been shown to out-compete both its parental taxa and produce monospecific stands. We surveyed the Pierce Cedar Creek Institute (PCCI) property for cattails and located 25 distinct cattail marshes. We determined the total area of cattail marsh at PCCI to be roughly 10% of the 267 ha property. Cattail individuals were sampled from each of the 25 stands and random amplified polymorphic DNA markers were used to identify the individuals to species. We found that 20 of the 25 stands were monospecific for the native cattail, T. latifolia. Five of the stands were mixtures of the native T. latifolia and the introduced T. angustifolia, and T. × glauca was found in two of the mixed stands. We recommend removal of the invasive T. angustifolia and T. × glauca individuals and the establishment of a monitoring plan in order to maintain the long-term health of the cattail marshes at PCCI. Keywords: Typha spp., RAPD markers, invasive species 1 INTRODUCTION Species of Typha L. (Typhaceae), commonly known as cattails, are highly productive emergent plants that grow in a variety of wetland habitats throughout the world (McManus et al. -

Federico Selvi a Critical Checklist of the Vascular Flora of Tuscan Maremma
Federico Selvi A critical checklist of the vascular flora of Tuscan Maremma (Grosseto province, Italy) Abstract Selvi, F.: A critical checklist of the vascular flora of Tuscan Maremma (Grosseto province, Italy). — Fl. Medit. 20: 47-139. 2010. — ISSN 1120-4052. The Tuscan Maremma is a historical region of central western Italy of remarkable ecological and landscape value, with a surface of about 4.420 km2 largely corresponding to the province of Grosseto. A critical inventory of the native and naturalized vascular plant species growing in this territory is here presented, based on over twenty years of author's collections and study of relevant herbarium materials and literature. The checklist includes 2.056 species and subspecies (excluding orchid hybrids), of which, however, 49 should be excluded, 67 need confirmation and 15 have most probably desappeared during the last century. Considering the 1.925 con- firmed taxa only, this area is home of about 25% of the Italian flora though representing only 1.5% of the national surface. The main phytogeographical features in terms of life-form distri- bution, chorological types, endemic species and taxa of particular conservation relevance are presented. Species not previously recorded from Tuscany are: Anthoxanthum ovatum Lag., Cardamine amporitana Sennen & Pau, Hieracium glaucinum Jord., H. maranzae (Murr & Zahn) Prain (H. neoplatyphyllum Gottschl.), H. murorum subsp. tenuiflorum (A.-T.) Schinz & R. Keller, H. vasconicum Martrin-Donos, Onobrychis arenaria (Kit.) DC., Typha domingensis (Pers.) Steud., Vicia loiseleurii (M. Bieb) Litv. and the exotic Oenothera speciosa Nutt. Key words: Flora, Phytogeography, Taxonomy, Tuscan Maremma. Introduction Inhabited by man since millennia and cradle of the Etruscan civilization, Maremma is a historical region of central-western Italy that stretches, in its broadest sense, from south- ern Tuscany to northern Latium in the provinces of Pisa, Livorno, Grosseto and Viterbo. -

Cattail, Typha Latifolia
BROAD-LEAVED the young shoots are steamed they taste like cabbage. The base of the stem where it attaches to the rhizome CATTAIL can be boiled or roasted like potatoes. The young flower stalks can be taken out of their sheaths and Typha latifolia L. can be boiled or steamed just like corn (Roos-Collins plant symbol = TYLA 1990; Clarke 1977). Contributed By: USDA, NRCS, National Plant Data Cattail pollen is a fine substitute for flours. It is a Center & Idaho Plant Materials Center bright yellow or green color, and turns pancakes, cookies or biscuits a pretty yellow color (which Alternate Names children love). The rhizomes (underground stems) Flags, rushes, bulrushes, cat o’nine tails, Cossack and lower stems have a sweet flavor and can be eaten raw, baked, roasted, or broiled. Cattail rhizomes are fairly high in starch content; this is usually listed at about 30% to 46%. The core can be ground into flour. One acre of cattails would yield about 6,475 pounds of flour (Harrington 1972). This flour would probably contain about 80 % carbohydrates and around 6% to 8% protein. Since cattail occurs around the world, it is a potential source of food for the worlds' population. Newly emerging shoots of cattails are edible, with delicate flavor and crispy asparagus like texture (Glenn Keator, Linda Yamane, Ann Lewis 1995). The end of a new stem of cattail is popular for eating with Washoes (Murphy 1959). When mixed with tallow, the brown fuzz can be chewed like gum. The Klamath and Modocs of northern California and southern Oregon make flexible baskets of twined tule or cattail. -

Aquatic Vascular Plants of New England, Station Bulletin, No.517
University of New Hampshire University of New Hampshire Scholars' Repository NHAES Bulletin New Hampshire Agricultural Experiment Station 2-1-1981 Aquatic vascular plants of New England, Station Bulletin, no.517 Crow, G. E. Hellquist, C. B. New Hampshire Agricultural Experiment Station Follow this and additional works at: https://scholars.unh.edu/agbulletin Recommended Citation Crow, G. E.; Hellquist, C. B.; and New Hampshire Agricultural Experiment Station, "Aquatic vascular plants of New England, Station Bulletin, no.517" (1981). NHAES Bulletin. 478. https://scholars.unh.edu/agbulletin/478 This Text is brought to you for free and open access by the New Hampshire Agricultural Experiment Station at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in NHAES Bulletin by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. S ON BULLETIN 517 February, 1981 oo-5n Aquatic Vascular Plants of Ne^v England: Part 2. Typhaceae and Sparganiaceae by G. E. Crow and C. B. Hellquist NEW HAMPSHIRE AGRICULTURAL EXPERIMENT STATION UNIVERSITY OF NEW HAMPSHIRE DURHAM, NEW HAMPSHIRE ON BULLETIN 517 February, 1981 Aquatic Vascular Plants of New England: Part 2. Typhaceae and Sparganiaceae by G. E. Crow and C. B. Hellquist NEW HAMPSHIRE AGRICULTURAL EXPERIMENT STATION UNIVERSITY OF NEW HAMPSHIRE DURHAM, NEW HAMPSHIRE •' Nst; Hrirr-poliire Y •'_v--r-r 5 %^ ACKNOWLEDGEMENTS ^^ 5/:^ We wish to thank Drs. Ernest O. Beal, Vernon L. Harms, Arthur C. Mathieson, and Eugene C. Ogden for their helpful comments on the manuscript. We are also grateful to the curators of the following her- baria for use of their collections: BOSC, BRU, CONN, CUW, GH, HNH, KIRI, MASS, MAINE, NCBS, NHA, NEBC, VT, YU. -

Narrowleaf Cattail
NARROWLEAF The Klamath and Modoc peoples of northern California and southern Oregon made flexible CATTAIL baskets of twined cattail. Cattails were also twined to form mats of varying sizes for sleeping, sitting, Typha angustifolia L. working, entertaining, covering doorways, providing plant symbol = TYAN shade, and a myriad of other uses. Lengths of cattail were plied into rope or other size cordage, and cattail Contributed By: USDA, NRCS, National Plant Data rope was used in some areas to bind bundles of tule Center & Idaho Plant Materials Center into tule boats. Air pockets or aerenchyma in the stems provided the buoyancy for good boat-building Alternate Names material. flags, rushes, bulrushes, cat o’nine The Cahuilla Indians used the stalks for matting, tails, Cossack bedding material, and ceremonial bundles (Barrows asparagus, reed 1967). Some tribes used the leaves and sheath bases mace, baco as caulking materials. Apaches used the pollen in female puberty ceremonies. After dipping the spike Uses in coal oil, the stalk makes a fine torch. The fluff can Caution: This also be used as tinder, insulation, or for lining baby species can be very cradleboards. The down is used for baby beds invasive in disturbed (Murphey 1959). wetlands. Please read about the Wildlife: The multitudes of tiny, wind-carried seeds environmental are too small and too hairy to be attractive to birds concerns under (Hotchkiss and Dozier 1949). In a few exceptions, Management. the seeds are eaten by several duck species. Cattail rootstocks are much more valuable as food for Ethnobotanic: All wildlife than are the seeds. Geese and muskrats parts of the cattail prefer the stems and roots. -

ELEMENT STEWARDSHIP ABSTRACT for Typha Spp. North
ELEMENT STEWARDSHIP ABSTRACT for Typha spp. North American Cattails To the User: Element Stewardship Abstracts (ESAs) are prepared to provide The Nature Conservancy's Stewardship staff and other land managers with current management-related information on those species and communities that are most important to protect, or most important to control. The abstracts organize and summarize data from numerous sources including literature and researchers and managers actively working with the species or community. We hope, by providing this abstract free of charge, to encourage users to contribute their information to the abstract. This sharing of information will benefit all land managers by ensuring the availability of an abstract that contains up-to-date information on management techniques and knowledgeable contacts. Contributors of information will be acknowledged within the abstract and receive updated editions. To contribute information, contact the editor whose address is listed at the end of the document. For ease of update and retrievability, the abstracts are stored on computer at the national office of The Nature Conservancy. This abstract is a compilation of available information and is not an endorsement of particular practices or products. Please do not remove this cover statement from the attached abstract. Authors of this Abstract: K. Motivans, S. Apfelbaum Applied Ecological Services, Inc © THE NATURE CONSERVANCY 1815 North Lynn Street, Arlington, Virginia 22209 (703) 841 5300 The Nature Conservancy Element Stewardship Abstract Typha spp North American cattails I. IDENTIFIERS Scientific-Name: Typha spp. Common-Name: cattail Description: The cattail genus (Typha spp.) is an erect, perennial freshwater aquatic herb which can grow 3 or more meters in height. -

Effects of Environmental Factors on Sparganium Emersum and Sparganium Erectum Colonization in Two Drainage Ditches with Different Maintenance
Vol.3, No.4, 538-544 (2012) Agricultural Sciences http://dx.doi.org/10.4236/as.2012.34064 Effects of environmental factors on Sparganium emersum and Sparganium erectum colonization in two drainage ditches with different maintenance Korehisa Kaneko1, Hiroshi Jinguji2 1Hokuso Creature Association, Tokyo, Japan; [email protected] 2School of Food, Agricultural and Environmental Sciences, Miyagi University, Sendai, Japan Received 12 March 2012; revised 25 April 2012; accepted 4 May 2012 ABSTRACT 1. INTRODUCTION In the Niheishimizu and Ooshimizu sections of [1] has established the “Ministry of Agriculture, For- the town of Misato in the Akita Prefecture, Nor- estry and Fisheries (MAFF) biodiversity strategy” to thern Japan, there are many abundant spring promote approaches to agriculture that recognise the water areas. Sparganium (Sparganium emersum value of biodiversity, reduce damage to the populations and Sparganium erectum) species are widely of local birds and other animals and protect agricultural distributed in the irrigation water that fed by resources. The MAFF is promoting types of environ- spring water. The irrigation waters were divided mentally friendly agriculture that can coexist with many the natural type ditch and the maintained ditch living organisms. It is necessary to maintain and restore rice fields, ditch and the habitats of wild flora and fauna. that connect with nearby natural ditch to pro- Spring water areas occur in some parts of the alluvial mote environmentally friendly agriculture. This fans located on the plains in the northern area of the study was conducted in both sections to sup- Senboku District in the Yokote basin, Akita Prefecture, port the maintenance of the irrigation water fed Northern Japan.