Report on the Deliberation Results June 7, 2017 Pharmaceutical
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

“Amenalief® Tab. 200Mg” in Japan
News Release July 3, 2017 Maruho Co., Ltd. Maruho receives manufacturing and marketing approval for Anti-Herpes Virus Agent “Amenalief® Tab. 200mg” in Japan Osaka (Japan), July 3, 2017 – Maruho Co., Ltd (“Maruho”, Head Office: Osaka, Japan, President and CEO: Koichi Takagi) announces that today it has received manufacturing and marketing approval from the Japanese Ministry of Health, Labor and Welfare (MHLW), for anti-herpes virus agent “Amenalief® Tab. 200mg” (INN: amenamevir) (hereinafter referred to as “the product”) for the treatment of herpes zoster (shingles) in Japan. The product is an anti-herpes virus agent with a novel mechanism of action created by Astellas Pharma Inc. (“Astellas”; Head Office: Tokyo, President and CEO: Yoshihiko Hatanaka). The product has been observed to inhibit the proliferation of the varicella-zoster virus (hereinafter VZV) by inhibiting the activity of the helicase-primase complex, which is essential for viral DNA replication. In August 2012, Maruho and Astellas agreed on a license agreement for the development and commercialization of the product in Japan, and Maruho has been progressing its development. Herpes zoster (shingles) is a disease caused by reactivation of the chickenpox VZV in latently-infected nerve ganglia. The main treatment for shingles is anti-herpes virus agents. The product is proven to be effective against VZV when administered once a day after meals. Also, since most of the product is excreted in feces, it is not necessary to adjust the dosage and administration according to creatinine clearance, an indicator of kidney function. Maruho hopes the product will contribute to the expansion of treatment options and improvement in adherence for the treatment of shingles in Japan. -
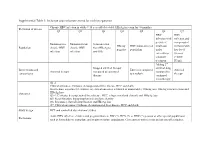
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

Herpes Simplex Virus
HSV Herpes simplex virus HSV (Herpes simplex virus) can be spread when an infected person is producing and shedding the virus. Herpes simplex can be spread through contact with saliva, such as sharing drinks. Symptoms of herpes simplex virus infection include watery blisters in the skin or mucous membranes of the mouth, lips or genitals. Lesions heal with ascab characteristic of herpetic disease. As neurotropic and neuroinvasive viruses, HSV-1 and -2 persist in the body by becoming latent and hiding from the immune system in the cell bodies of neurons. After the initial or primary infection, some infected people experience sporadic episodes of viral reactivation or outbreaks. www.MedChemExpress.com 1 HSV Inhibitors (Z)-Capsaicin 1-Docosanol (Zucapsaicin; Civamide; cis-Capsaicin) Cat. No.: HY-B1583 (Behenyl alcohol) Cat. No.: HY-B0222 (Z)-Capsaicin is the cis isomer of capsaicin, acts 1-Docosanol is a saturated fatty alcohol used as an orally active TRPV1 agonist, and is used in traditionally as an emollient, emulsifier, and the research of neuropathic pain. thickener in cosmetics, and nutritional supplement; inhibitor of lipid-enveloped viruses including herpes simplex. Purity: 99.96% Purity: ≥98.0% Clinical Data: Launched Clinical Data: Launched Size: 10 mM × 1 mL, 10 mg, 50 mg Size: 500 mg 2-Deoxy-D-glucose 20(R)-Ginsenoside Rh2 (2-DG; 2-Deoxy-D-arabino-hexose; D-Arabino-2-deoxyhexose) Cat. No.: HY-13966 Cat. No.: HY-N1401 2-Deoxy-D-glucose is a glucose analog that acts as 20(R)-Ginsenoside Rh2, a matrix a competitive inhibitor of glucose metabolism, metalloproteinase (MMP) inhibitor, acts as a inhibiting glycolysis via its actions on hexokinase. -

WHO Drug Information Vol
WHO Drug Information Vol. 33, No. 2, 2019 WHO Drug Information Contents Publication News 135 53rd report of the World Health Organization (WHO) Expert Committee on Specifications for Pharmaceutical Preparations (ECSPP) Consultation Documents 139 Concept Note: A Framework for Evaluating and Publicly Designating Regulatory Authorities as WHO-Listed Authorities 159 Quality Management System Requirements for National Inspectorates 174 Procedure for the Elaboration, Revision and Omission of Monographs and Other Texts for The International Pharmacopoeia 179 Good Chromatography Practices 194 Good Storage and Distribution Practices for Medical Products ATC/DD Classification 226 ATC/DDD Classification (Temporary) 229 ATC/DDD Classification (Final) International Nonproprietary Names (INN) 233 List No. 121 of Proposed International Nonproprietary Names (INN) for Pharmaceutical Substances Abbreviations and websites CHMP Committee for Medicinal Products for Human Use (EMA) EMA European Medicines Agency (www.ema.europa.eu) EU European Union FDA U.S. Food and Drug Administration (www.fda.gov) Health Canada Federal department responsible for health product regulation in Canada (www.hc-sc.gc.ca) HPRA Health Products Regulatory Authority, Ireland (www.hpra.ie) HSA Health Sciences Authority, Singapore (www.hsa.gov.sg) ICDRA International Conference of Drug Regulatory Authorities ICH International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (www.ich.org) IGDRP International Generic Drug Regulators Programme (https://www.igdrp.com) -
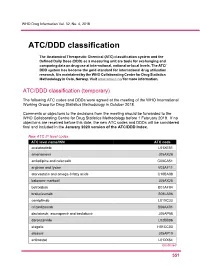
ATC/DDD Classification
WHO Drug Information Vol. 32, No. 4, 2018 ATC/DDD classification The Anatomical Therapeutic Chemical (ATC) classification system and the Defined Daily Dose (DDD) as a measuring unit are tools for exchanging and comparing data on drug use at international, national or local levels. The ATC/ DDD system has become the gold standard for international drug utilization research. It is maintained by the WHO Collaborating Centre for Drug Statistics Methodology in Oslo, Norway. Visit www.whocc.no/ for more information. ATC/DDD classification (temporary) The following ATC codes and DDDs were agreed at the meeting of the WHO International Working Group for Drug Statistics Methodology in October 2018. Comments or objections to the decisions from the meeting should be forwarded to the WHO Collaborating Centre for Drug Statistics Methodology before 1 February 2019. If no objections are received before this date, the new ATC codes and DDDs will be considered final and included in the January 2020 version of the ATC/DDD Index. New ATC 5th level codes ATC level name/INN ATC code acalabrutinib L01XE51 amenamevir J05AX26 amlodipine and celecoxib C08CA51 arginine and lysine V03AF11 atorvastatin and omega-3 fatty acids C10BA08 baloxavir marboxil J05AX25 betrixaban B01AF04 brolucizumab S01LA06 cemiplimab L01XC33 crizanlizumab B06AX01 daclatasvir, asunaprevir and beclabuvir J05AP58 darolutamide L02BB06 elagolix H01CC03 elbasvir J05AP10 entinostat L01XX64 Continued 5 51 ATC/DDD classification WHO Drug Information Vol. 32, No. 4, 2018 New ATC 5th level codes (continued) -

Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2
viruses Review Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2 Lauren A. Sadowski 1,†, Rista Upadhyay 1,2,†, Zachary W. Greeley 1,‡ and Barry J. Margulies 1,3,* 1 Towson University Herpes Virus Lab, Department of Biological Sciences, Towson University, Towson, MD 21252, USA; [email protected] (L.A.S.); [email protected] (R.U.); [email protected] (Z.W.G.) 2 Towson University Department of Chemistry, Towson, MD 21252, USA 3 Molecular Biology, Biochemistry, and Bioinformatics Program, Towson University, Towson, MD 21252, USA * Correspondence: [email protected] † Authors contributed equally to this manuscript. ‡ Current address: Becton-Dickinson, Sparks, MD 21152, USA. Abstract: Herpes simplex viruses-1 and -2 (HSV-1 and -2) are two of the three human alphaher- pesviruses that cause infections worldwide. Since both viruses can be acquired in the absence of visible signs and symptoms, yet still result in lifelong infection, it is imperative that we provide interventions to keep them at bay, especially in immunocompromised patients. While numerous experimental vaccines are under consideration, current intervention consists solely of antiviral chemotherapeutic agents. This review explores all of the clinically approved drugs used to prevent the worst sequelae of recurrent outbreaks by these viruses. Keywords: acyclovir; ganciclovir; cidofovir; vidarabine; foscarnet; amenamevir; docosanol; nelfi- navir; HSV-1; HSV-2 Citation: Sadowski, L.A.; Upadhyay, R.; Greeley, Z.W.; Margulies, B.J. Current Drugs to Treat Infections 1. Introduction with Herpes Simplex Viruses-1 and -2. The world of anti-herpes simplex (anti-HSV) agents took flight in 1962 with the FDA Viruses 2021, 13, 1228. -

Influenza Sampler
Influenza Sampler Presenting sample chapters on influenza from the Manual of Clinical Microbiology, 12th Edition, Chapter 86 “Influenza Viruses” by Robert L. Atmar This chapter discusses seasonal influenza strains as well as novel swine and avian influenza strains that can infect people and have pandemic potential. Chapter 83 “Algorithms for Detection and Identification of Viruses” by Marie Louise Landry, Angela M. Caliendo, Christine C. Ginocchio, Randall Hayden, and Yi-Wei Tang This chapter outlines technological advances for the diagnosis of viral infections. Chapter 113 “Antiviral Agents” by Carlos A.Q. Santos and Nell S. Lurian This chapter reviews antiviral agents approved by FDA and their mechanism(s) of action. Photo Credit: CDC/ Douglas Jordan, Dan Higgins Influenza Viruses* ROBERT L. ATMAR Send proofs to: Robert L. Atmar Email: [email protected] and to editors: [email protected] [email protected] [email protected] 86 TAXONOMY The segmented genome of influenza viruses allows the The influenza viruses are members of the family Orthomyxo- exchange of one or more gene segments between two viruses viridae. Antigenic differences in two major structural pro- when both infect a single cell. This exchange is called teins, the matrix protein (M) and the nucleoprotein (NP), genetic reassortment and results in the generation of new and phylogenetic analyses of the virus genome are used to strains containing a mix of genes from both parental viruses. separate the influenza viruses into four genera within the Genetic reassortment between human and avian influenza family: Influenzavirus A, Influenzavirus B, Influenzavirus C, virus strains led to the generation of the 1957 H2N2 and and Influenzavirus D. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. -

A Abacavir Abacavirum Abakaviiri Abagovomab Abagovomabum
A abacavir abacavirum abakaviiri abagovomab abagovomabum abagovomabi abamectin abamectinum abamektiini abametapir abametapirum abametapiiri abanoquil abanoquilum abanokiili abaperidone abaperidonum abaperidoni abarelix abarelixum abareliksi abatacept abataceptum abatasepti abciximab abciximabum absiksimabi abecarnil abecarnilum abekarniili abediterol abediterolum abediteroli abetimus abetimusum abetimuusi abexinostat abexinostatum abeksinostaatti abicipar pegol abiciparum pegolum abisipaaripegoli abiraterone abirateronum abirateroni abitesartan abitesartanum abitesartaani ablukast ablukastum ablukasti abrilumab abrilumabum abrilumabi abrineurin abrineurinum abrineuriini abunidazol abunidazolum abunidatsoli acadesine acadesinum akadesiini acamprosate acamprosatum akamprosaatti acarbose acarbosum akarboosi acebrochol acebrocholum asebrokoli aceburic acid acidum aceburicum asebuurihappo acebutolol acebutololum asebutololi acecainide acecainidum asekainidi acecarbromal acecarbromalum asekarbromaali aceclidine aceclidinum aseklidiini aceclofenac aceclofenacum aseklofenaakki acedapsone acedapsonum asedapsoni acediasulfone sodium acediasulfonum natricum asediasulfoninatrium acefluranol acefluranolum asefluranoli acefurtiamine acefurtiaminum asefurtiamiini acefylline clofibrol acefyllinum clofibrolum asefylliiniklofibroli acefylline piperazine acefyllinum piperazinum asefylliinipiperatsiini aceglatone aceglatonum aseglatoni aceglutamide aceglutamidum aseglutamidi acemannan acemannanum asemannaani acemetacin acemetacinum asemetasiini aceneuramic -

Advances and Perspectives in the Management of Varicella-Zoster Virus Infections
molecules Review Advances and Perspectives in the Management of Varicella-Zoster Virus Infections Graciela Andrei * and Robert Snoeck Laboratory of Virology and Chemotherapy, Rega Institute for Medical Research, KU Leuven, 3000 Leuven, Belgium; [email protected] * Correspondence: [email protected]; Tel.: +32-16-32-19-51 Abstract: Varicella-zoster virus (VZV), a common and ubiquitous human-restricted pathogen, causes a primary infection (varicella or chickenpox) followed by establishment of latency in sensory ganglia. The virus can reactivate, causing herpes zoster (HZ, shingles) and leading to significant morbidity but rarely mortality, although in immunocompromised hosts, VZV can cause severe disseminated and occasionally fatal disease. We discuss VZV diseases and the decrease in their incidence due to the introduction of live-attenuated vaccines to prevent varicella or HZ. We also focus on acyclovir, valacyclovir, and famciclovir (FDA approved drugs to treat VZV infections), brivudine (used in some European countries) and amenamevir (a helicase-primase inhibitor, approved in Japan) that augur the beginning of a new era of anti-VZV therapy. Valnivudine hydrochloride (FV-100) and valomaciclovir stearate (in advanced stage of development) and several new molecules potentially good as anti-VZV candidates described during the last year are examined. We reflect on the role of antiviral agents in the treatment of VZV-associated diseases, as a large percentage of the at-risk population is not immunized, and on the limitations of currently FDA-approved anti-VZV drugs. Their low efficacy in controlling HZ pain and post-herpetic neuralgia development, and the need of Citation: Andrei, G.; Snoeck, R. multiple dosing regimens requiring daily dose adaptation for patients with renal failure urges the Advances and Perspectives in the development of novel anti-VZV drugs. -

Repurposing of FDA Approved Drugs
Antiviral Drugs (In Phase IV) ABACAVIR GEMCITABINE ABACAVIR SULFATE GEMCITABINE HYDROCHLORIDE ACYCLOVIR GLECAPREVIR ACYCLOVIR SODIUM GRAZOPREVIR ADEFOVIR DIPIVOXIL IDOXURIDINE AMANTADINE IMIQUIMOD AMANTADINE HYDROCHLORIDE INDINAVIR AMPRENAVIR INDINAVIR SULFATE ATAZANAVIR LAMIVUDINE ATAZANAVIR SULFATE LEDIPASVIR BALOXAVIR MARBOXIL LETERMOVIR BICTEGRAVIR LOPINAVIR BICTEGRAVIR SODIUM MARAVIROC BOCEPREVIR MEMANTINE CAPECITABINE MEMANTINE HYDROCHLORIDE CARBARIL NELFINAVIR CIDOFOVIR NELFINAVIR MESYLATE CYTARABINE NEVIRAPINE DACLATASVIR OMBITASVIR DACLATASVIR DIHYDROCHLORIDE OSELTAMIVIR DARUNAVIR OSELTAMIVIR PHOSPHATE DARUNAVIR ETHANOLATE PARITAPREVIR DASABUVIR PENCICLOVIR DASABUVIR SODIUM PERAMIVIR DECITABINE PERAMIVIR DELAVIRDINE PIBRENTASVIR DELAVIRDINE MESYLATE PODOFILOX DIDANOSINE RALTEGRAVIR DOCOSANOL RALTEGRAVIR POTASSIUM DOLUTEGRAVIR RIBAVIRIN DOLUTEGRAVIR SODIUM RILPIVIRINE DORAVIRINE RILPIVIRINE HYDROCHLORIDE EFAVIRENZ RIMANTADINE ELBASVIR RIMANTADINE HYDROCHLORIDE ELVITEGRAVIR RITONAVIR EMTRICITABINE SAQUINAVIR ENTECAVIR SAQUINAVIR MESYLATE ETRAVIRINE SIMEPREVIR FAMCICLOVIR SIMEPREVIR SODIUM FLOXURIDINE SOFOSBUVIR FOSAMPRENAVIR SORIVUDINE FOSAMPRENAVIR CALCIUM STAVUDINE FOSCARNET TECOVIRIMAT FOSCARNET SODIUM TELBIVUDINE GANCICLOVIR TENOFOVIR ALAFENAMIDE GANCICLOVIR SODIUM TENOFOVIR ALAFENAMIDE FUMARATE TIPRANAVIR VELPATASVIR TRIFLURIDINE VIDARABINE VALACYCLOVIR VOXILAPREVIR VALACYCLOVIR HYDROCHLORIDE ZALCITABINE VALGANCICLOVIR ZANAMIVIR VALGANCICLOVIR HYDROCHLORIDE ZIDOVUDINE Antiviral Drugs (In Phase III) ADEFOVIR LANINAMIVIR OCTANOATE -

WHO Pharmaceuticals Newsletters
2020 WHO Pharmaceuticals No.3 NEWSLETTER The WHO Pharmaceuticals Newsletter provides you with the latest information on the safety of medicines WHO Vision for Medicines Safety and legal actions taken by regulatory authorities around No country left behind: the world. It also provides signals based on information worldwide pharmacovigilance for safer medicines, safer patients derived from the WHO global database of individual case safety reports, VigiBase. This edition of the Newsletter also includes three short The aim of the Newsletter is to disseminate regulatory articles, on: the WHO Vaccine Safety Net, a working information on the safety of group meeting on the ‘Erice call for change’ and the pharmaceutical products, launch of the Med Safety mobile application in based on communications received from our network of Uganda. national pharmacovigilance centres and other sources such as specialized bulletins and journals, as well as partners in WHO. The information is produced in the form of résumés in English, full texts of which may be obtained on request from: Safety and Vigilance: Medicines, EMP-HIS, World Health Organization, 1211 Geneva 27, Switzerland, Contents E -mail address: [email protected] This Newsletter is also available at: Regulatory matters http://www.who.int/medicines Safety of medicines Signal Feature WHO Pharmaceuticals Newsletter No. 3, 2020 ISBN 978-92-4-000726-0 (electronic version) ISBN 978-92-4-000727-7 (print version) © World Health Organization 2020 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo).