Olefin Polymerization Over Supported Chromium Oxide Catalysts
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

© Copyright 2013 Jennifer L. Steele
© Copyright 2013 Jennifer L. Steele DIVALENT TRANSITION METAL CENTERS: THE SYNTHESIS OF NEW CHEMICAL VAPOR DEPOSITION PRECURSORS AND STUDIES OF ETHYLENE POLYMERIZATION AND OLIGOMERIZATION CATALYSTS BY JENNIFER L. STEELE DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate College of the University of Illinois at Urbana-Champaign, 2013 Urbana, Illinois Doctoral Committee: Professor Gregory S. Girolami, Chair Assistant Professor Alison R. Fout Professor John A. Katzenellenbogen Professor Thomas B. Rauchfuss Abstract Volatile transition metal complexes that contain boron hydride ligands are desirable for their potential as precursors for metal diboride films for microelectronics applications. Recently our group has discovered a new class of potential precursors in the metal complexes of the chelating borohydride, N,N-dimethylaminodiboranate (DMADB). To date, attempts to synthesize homoleptic complexes of the late transition metals have afforded intractable mixtures, likely the result of overreduction of the metal center. This work has focused on the synthesis and characterization of heteroleptic complexes of the late transition metals that contain both DMADB and 1,2,3,4,5,-pentamethylcyclopentadienyl ligands. The reaction of metal complexes of the form [Cp*MX]n, where Cp* is 1,2,3,4,5,- pentamethylcyclopentadienyl, M = Cr, Fe, Co, or Ru, and X = Cl or I with sodium dimethylaminodiboranate (NaDMADB) in diethyl ether affords the divalent complexes [Cp*M(DMADB)]. Additionally, the analogous vanadium compound [Cp*V(DMADB)] can be synthesized from the reduction of [Cp*VCl2]3 with NaDMADB in diethyl ether. All of these compounds are volatile under static vacuum at room temperature, but are also thermally sensitive; the iron and ruthenium derivatives decompose at room temperature over a day. -

Chapter 2 Polymerisation of MMA Using Novel Chromium(II)
A Thesis Submitted for the Degree of PhD at the University of Warwick Permanent WRAP URL: http://wrap.warwick.ac.uk/133954 Copyright and reuse: This thesis is made available online and is protected by original copyright. Please scroll down to view the document itself. Please refer to the repository record for this item for information to help you to cite it. Our policy information is available from the repository home page. For more information, please contact the WRAP Team at: [email protected] warwick.ac.uk/lib-publications Novel chromium compounds and their use in the polymerisation of methyl methacrylate Mark Andrew Stump A Thesis submitted for the Degree of Doctor of Philosophy Department of Chemistry University of Warwick Coventry CV4 7AL 18th January 1998 A: Table of Contents A Table of Contents 1• B Table of Tables Vlll• • • C Table of Figures xiv D Declaration xix E Acknowledgements XX F Summary xxi G Abbreviations xxii 1. Introduction 2 1.1. An Introduction to Polymers 12 3 1.2. Types of Polymerisation 5 1.2.1. Anionic Polymerisation 5 1.2.2. Co-ordination Polymerisation 7 1.2.3. Radical Polymerisation 9 1.3. Controlled Polymerisation 10 1.3.1. Stereochemistry of vinyl polymerisation 10 1.3.2. Other Aspects of Controlled Polymerisation 12 1.4. Living Polymerisation 13 1.5. An Introduction To Chromium Chemistry 22 1.6. Methods of Analysis for Polymers 34 1.6.1. Size Exclusion Chromatography (SEC) 34 1.6.2. Thermal Gravimetric Analysis (TGA) 35 1.7. References 44 2. Polymerisation of MMA using novel chromium(II) and (III) compounds in conjunction with alkyl halides 49 2.1. -

Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc
Metal-Metal (MM) Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc Richard H. Duncan Lyngdoh*,a, Henry F. Schaefer III*,b and R. Bruce King*,b a Department of Chemistry, North-Eastern Hill University, Shillong 793022, India B Centre for Computational Quantum Chemistry, University of Georgia, Athens GA 30602 ABSTRACT: This survey of metal-metal (MM) bond distances in binuclear complexes of the first row 3d-block elements reviews experimental and computational research on a wide range of such systems. The metals surveyed are titanium, vanadium, chromium, manganese, iron, cobalt, nickel, copper, and zinc, representing the only comprehensive presentation of such results to date. Factors impacting MM bond lengths that are discussed here include (a) n+ the formal MM bond order, (b) size of the metal ion present in the bimetallic core (M2) , (c) the metal oxidation state, (d) effects of ligand basicity, coordination mode and number, and (e) steric effects of bulky ligands. Correlations between experimental and computational findings are examined wherever possible, often yielding good agreement for MM bond lengths. The formal bond order provides a key basis for assessing experimental and computationally derived MM bond lengths. The effects of change in the metal upon MM bond length ranges in binuclear complexes suggest trends for single, double, triple, and quadruple MM bonds which are related to the available information on metal atomic radii. It emerges that while specific factors for a limited range of complexes are found to have their expected impact in many cases, the assessment of the net effect of these factors is challenging. -
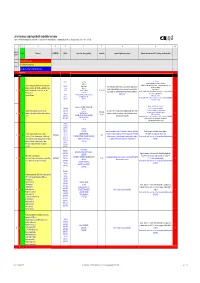
List of Substances Subject to Prohibited / Undesirable / Declaration
List of substances subject to prohibited / undesirable / declaration Appendix 1 to the Guidelines to Design and Purchasing Prohibition of Harmful Substances : ebmpapstMulfingen: 90-001 ebmpapstSt.Georgen: WN 3-12.2 ebmpapstLandshut: 60118.00040 (Ver. 5.0-31.07.2008) 1 2 3 4 5 6 7 8 9 10 Consecu N - new Substance EU-INDEX-No.: CAS-No. Source (law, decree, guideline). Hazard/risk example of application/occurrence Remarks and comments (of 0.1% deviating consideration limit) tive No. V = prohibited (verboten) U = undesirable (unerwünscht) D = subject to declaration (deklarationspflichtig) V = prohibited RoHS from 100 ppm 7439-92-1 TRGS 905, Pb-stabilisers and pigments are subject to declaration. GefStoffV, Prohibited: Anhydrite neutral lead carbonate, lead hydrogen carbonate and lead Lead and compounds (red lead oxide, lead sulphate, lead (7446-14-2 ChemVerbotsV, cable coating, heat transfer medium for accumulators, hybrid integrated sulphate as dye pigment hydrogen carbonate, lead carbonate, lead phtalates, lead 1319-46-6 BatterieV, circuits, stabilisers in plastics, vessels and pipes for aggressive liquids, < 0.4 % in batteries 1 V N acetates, lead phosphates, lead stereates , etc) lead 598-63-0 2002/95/EG (RoHS), K3, R 1, R 3 V E F according to RoHS: Lead as alloying additive in aluminum (up to 0.4 mass%) in steel (up to 67/458/EEC pigment production, lead alloys, anti-corrosion agents (fuel additives), chromate: refer to 0.35 mass%), in copper (up to 4%), 301-04-2 76/769/EEC (89/667/EEC, 97/10/EC, 97/56/EC) soldering agent chromium (VI) -

Sd0000029 Evaluation of Chromfte Ore and The
SD0000029 EVALUATION OF CHROMFTE ORE AND THE OPTIMUM METHODS FOR INDUSTRIAL EXTRACTION OF CHROMIUM A TI1HSIS SUBMITTED BY Bakheit Mustafa Mohamed Salih IN CANDIDATURE FOR TIU:DI:GRFJ:OI MASTER OF SC1HNCT DEPARTMENT OF CHEMISTRY FACULTY OF SCIENCE (P.O.BOX 321) OCTOBER 1999 UNIVERSITY OF KHARTOUM 31/ 28 ABSTRACT Samples of chromite ore, collected from Gam and Cheikay mining area (Ingessana Hills) in east Sudan, were analysed to assess the chromium content. Methods for extraction and analysis of chromium metal were developed and established. Analysis were carried out using atomic absorption spectroscopy (AAS) to estimate the contents of chromium, iron, calcium, and magnesium. X-ray fluorescence (XRF) was used to evaluate the levels of chromium, iron, and calcium in the ore. Volumetric analysis was performed to assess chromium and iron, whilst gravimetric analysis was employed to measure the amounts of calcium, magnesium, aluminum and silicon present in the ore. The data was chemically and statistically analyzed to compare the results obtained by the given analytical methods. The results are in good agreement except iron oxide, which displayed a significantly different value when measured by x-ray fluorescence. The data obtained exhibited similarity in almost all cases, when compared with local and global researches, reports, and literature. The study has revealed the average contents of Cr2O3, FeO, CaO, MgO, A12O3, and SiO2 as 40.66. 11.96, 11.94. 0.36. 16.94, 11.45% respectively. MnO and NiO were detected in trace amounts, the corresponding levels in the ore being 72 and 27 ppm. The average chromium content in extracted potassium dichromate measured by using AAS, XRF, and volumetric methods was found to be 31.7%. -

Potassium Dichromate CAS Number: 7778-50-9 EC Number: 231-906-6
21.5.2010 COMMENTS AND RESPONSE TO COMMENTS ON ANNEX XV SVHC: PROPOSAL AND JUSTIFICATION Substance name: Potassium dichromate CAS number: 7778-50-9 EC number: 231-906-6 Reason of the submission of the Annex XV: CMR Disclaimer: The European Chemicals Agency is not responsible for the content of this document. The Response to Comments table has been prepared by the competent authority of the Member State preparing the proposal for identification of a Substance of Very High Concern. The comments were received during the public consultation of the Annex XV dossier. General comments Date Submitted by (name, Comment Response Organisation/MSCA) 20100323 CLEAPSS, UK (on behalf of an CLEAPSS is an advisory service providing support in No answer expected. organisation) science and technology for a consortium of local authorities and their schools including establishments for pupils with special needs. Independent schools, post-16 colleges, teacher training establishments, curriculum developers and others can apply for associate membership. We offer help from nursery education through to A-level studies or equivalent. Our services cover health & safety, risk assessment, sources and use of chemicals, living organisms and equipment. CLEAPSS also provides advice on technicians & their jobs, as well as the design of laboratories and facilities & fittings for D&T and science rooms. 20100324 Germany, company (on behalf of an We reject the ban of Potassium Dichromate in general, Thank for your comment. organisation) because the substance is used in a low concentration (< 1%) in an almost closed system of cuvette tests 20100408 Draeger Safety AG & Co. KGaA, comments k2Cr2O7R1.doc Thank you for this information. -

Rpt POL-TOXIC AIR POLLUTANTS 98 BY
SWCAA TOXIC AIR POLLUTANTS '98 by CAS ASIL TAP SQER CAS No HAP POLLUTANT NAME HAP CAT 24hr ug/m3 Ann ug/m3 Class lbs/yr lbs/hr none17 BN 1750 0.20 ALUMINUM compounds none0.00023 AY None None ARSENIC compounds (E649418) ARSENIC COMPOUNDS none0.12 AY 20 None BENZENE, TOLUENE, ETHYLBENZENE, XYLENES BENZENE none0.12 AY 20 None BTEX BENZENE none0.000083 AY None None CHROMIUM (VI) compounds CHROMIUM COMPOUN none0.000083 AY None None CHROMIUM compounds (E649962) CHROMIUM COMPOUN none0.0016 AY 0.5 None COKE OVEN COMPOUNDS (E649830) - CAA 112B COKE OVEN EMISSIONS none3.3 BN 175 0.02 COPPER compounds none0.67 BN 175 0.02 COTTON DUST (raw) none17 BY 1,750 0.20 CYANIDE compounds CYANIDE COMPOUNDS none33 BN 5,250 0.60 FIBROUS GLASS DUST none33 BY 5,250 0.60 FINE MINERAL FIBERS FINE MINERAL FIBERS none8.3 BN 175 0.20 FLUORIDES, as F, containing fluoride, NOS none0.00000003 AY None None FURANS, NITRO- DIOXINS/FURANS none5900 BY 43,748 5.0 HEXANE, other isomers none3.3 BN 175 0.02 IRON SALTS, soluble as Fe none00 AN None None ISOPROPYL OILS none0.5 AY None None LEAD compounds (E650002) LEAD COMPOUNDS none0.4 BY 175 0.02 MANGANESE compounds (E650010) MANGANESE COMPOU none0.33 BY 175 0.02 MERCURY compounds (E650028) MERCURY COMPOUND none33 BY 5,250 0.60 MINERAL FIBERS ((fine), incl glass, glass wool, rock wool, slag w FINE MINERAL FIBERS none0.0021 AY 0.5 None NICKEL 59 (NY059280) NICKEL COMPOUNDS none0.0021 AY 0.5 None NICKEL compounds (E650036) NICKEL COMPOUNDS none0.00000003 AY None None NITROFURANS (nitrofurans furazolidone) DIOXINS/FURANS none0.0013 -

Wavelength Dependence of Photooxidation Vs Photofragmentation of Chromocene
J. Phys. Chem. A 2001, 105, 8665-8671 8665 Wavelength Dependence of Photooxidation vs Photofragmentation of Chromocene Peter T. Muraoka, Daniel Byun, and Jeffrey I. Zink* Department of Chemistry and Biochemistry, UniVersity of California, Los Angeles, California 90095 ReceiVed: March 19, 2001; In Final Form: July 2, 2001 Photooxidation and metal-ligand photolysis reactions of bis(cyclopentadienyl)chromium, chromocene, in the range 24 390-15 630 cm-1 are studied in the gas phase by using time-of-flight mass spectroscopic detection. Photooxidation of the intact chromocene molecule unexpectedly dominates in the range 23 530-24 000 cm-1. The relative importance of photooxidation compared to photofragmentation is strongly wavelength dependent. A prominent species at all wavelengths is the chromium ion, but in a wavelength region corresponding to the lowest energy ligand to metal charge transfer excited electronic state absorption, the strongest peak is from the chromocene ion. The excitation spectra are reported for three selected species: chromocene ion, mono- (cyclopentadienyl)chromium ion, and the chromium ion. The spectrum obtained by monitoring the metal ion contains sharp peaks that are assigned to neutral chromium atom resonances. Sharp losses of intensities in the molecular ion spectra are observed at these wavelengths. The wavelength dependencies of the photoreactions are interpreted and explained in terms of the identity of the initially populated excited electronic state and the ionization energy of the molecule. When the initially populated excited electronic state is the ligand to metal charge transfer state, the first photon causes minimal bond weakening and the second photon excites the intact chromocene above the ionization energy, resulting in efficient ionization of the parent molecule. -

Chromate and the Environment: Removal and Utilization of Industrial Waste
J. Chem. Chem. Eng. 10 (2016) 147-152 doi: 10.17265/1934-7375/2016.03.006 D DAVID PUBLISHING Chromate and the Environment: Removal and Utilization of Industrial Waste Fernando B. Mainier, Pedro Paulo B. Leite, Marcone F. Reis and Thiago Teobaldo Silva Engineering School, Federal Fluminense University(UFF), Niterói, Rio de Janeiro 24220-261, Brazil Abstract: Chromate and dichromate sodium as a function of oxidizer characteristics are used in several industrial areas; for example, in surface protection of coated parts of cadmium, zinc and aluminum (chromate coated treated), corrosion inhibitors, the treatment of leather, the manufacture of pigments, etc. However, the use of such products has been questioned due to the problems of toxicity and pollution that can be caused in the environmental. The Brazilian environmental agency has established that the concentrations of 2- chromate in water courses are less than 0.5 ppm. In order to reuse chromate (CrO4 ) from industrial effluent, laboratory experiments have been proposed based on chemical reduction or electrolytic processes, in order to transform these chromate ions in a final mix of oxides (in solid form), which can then be packed and sent to the production process of sodium chromate. The results of these experiments have become useful industrially (without regard to costs) considering the environmental reuse and the life cycle of the chemical compound. Key words: Chromate, dichromate, contamination, chemical reduction, electrolytic process. 1. Introduction waste, among others [1, 2]. In -

Non-Bridged Half-Metallocene Complexes of Group 4–6 Metals with Chelating Ligands As Well-Defined Catalysts for A-Olefin Polymerization
Polymer Journal (2015) 47, 2–17 & 2015 The Society of Polymer Science, Japan (SPSJ) All rights reserved 0032-3896/15 www.nature.com/pj INVITED REVIEW Non-bridged half-metallocene complexes of group 4–6 metals with chelating ligands as well-defined catalysts for a-olefin polymerization Hayato Tsurugi, Keishi Yamamoto, Raphae¨l Rochat and Kazushi Mashima Tremendous effort has been directed toward the design of an organic ligand framework around the catalytically active metal center of homogeneous catalyst precursors. This work is aimed at controlling not only the reactivity of the metal catalysts for a-olefin polymerization but also the molecular weight, molecular weight distribution, polymer microstructure and monomer content of the copolymers. Among the catalyst precursor categories, non-bridged half-metallocene complexes supported by a variety of chelating ligands are attractive catalyst motifs for ethylene homopolymerization, ethylene/a-olefin copolymerization and stereoselective polymerization of a-olefins. These motifs are attractive in terms of their rather simple synthetic protocols and the wide range of potential architectural designs of the attached ligands. This review article summarizes recent developments regarding non-bridged half-metallocene complexes of group 4–6 metals with anionic chelating ligands. In 5 contrast to the conventional metallocene initiators Cp2MX2 (Cp ¼ g -C5H5), half-metallocene complexes of the type CpM(L^L)X2 (L^L ¼ chelating ligands) offer the advantage of catalyst modification. Steric and/or electronic modification of the coordination environment can be achieved by changing one cyclopentadienyl ligand of a metallocene complex to another ligand, such as three-, four-, five-, six- or seven-membered chelates having bidentate or tridentate coordinations and being monoanionic, dianionic or trianionic. -

Electrochemistry Evaluation of Chromocene in Organic Solvents for Non-Aqueous Organic Redox Flow Electrolyte
International Proceedings of Chemical, Biological and Environmental Engineering, V0l. 102 (2017) DOI: 10.7763/IPCBEE. 2017. V102. 3 Electrochemistry Evaluation of Chromocene in Organic Solvents for Non-Aqueous Organic Redox Flow Electrolyte Yongbeom Kim 1, Youngho Lee 2 and Joonhyeon Jeon 1 1 Department of Electronic & Electrical engineering, Dongguk University, Pildongro-1gil street, Seoul, Republic of Korea 2 Department of Energy and Advanced Material Engineering, Dongguk University, Pildongro-1gil street, Seoul, Republic of Korea Abstract. The redox flow battery (RFB) is kind of energy storage device for large scale energy storage system. It has many advantages for large scale energy storage system. But, conventional electrolyte has low energy density. So, for overcoming this problem, the chromocene, which is kind of metallocene and has high standard voltage, is studied for RFB. In this paper, the solvents (1-dioxolane, 2-tetrahydrofuran, 3-N,N- dimethylformamaide, 4-benzene, 5-hexane, 6-toluene, 7-heptane, 8-acetotrile, 9-propylene carbonate, 10-N- methyle-2-pyrrolidinone) for chromocene is researched. Solubility, electrical conductivity, electrochemistry properties determined by many experiments. As a result, N-metyle-2-pyrrolidinone has highest conductivity and toluene has highest solubility for using chromocene. Also, N-metyle-2-pyrrolidinone has suitable redox reaction and electrochemical property for RFB. Synthetically, N-metyle-2-pyrrolidinone is determined that it is proper for solvent of chromocene in RFB. Keywords: redox couple, chromocene, metallocene, non-aqueous electrolyte, flow battery 1. Introduction Recently, as increasing demand of electricity and renewable energy, Energy storage devices are interested by industry and researchers. Many kind of Energy storage device (Li-ion battery [1], super capacitor [2], redox flow battery, [3]-[5] Sodium-sulfur battery [6], etc.) are compete for energy storage market. -

The Elements.Pdf
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 3) [5/17/2001 4:06:20 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions.