A QM-CAMD Approach to Solvent Design for Optimal Reaction Rates
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Vibrationally Excited Hydrogen Halides : a Bibliography On
VI NBS SPECIAL PUBLICATION 392 J U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards National Bureau of Standards Bldg. Library, _ E-01 Admin. OCT 1 1981 191023 / oO Vibrationally Excited Hydrogen Halides: A Bibliography on Chemical Kinetics of Chemiexcitation and Energy Transfer Processes (1958 through 1973) QC 100 • 1X57 no. 2te c l !14 c '- — | NATIONAL BUREAU OF STANDARDS The National Bureau of Standards' was established by an act of Congress March 3, 1901. The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau consists of the Institute for Basic Standards, the Institute for Materials Research, the Institute for Applied Technology, the Institute for Computer Sciences and Technology, and the Office for Information Programs. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consistent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry, and commerce. The Institute consists of a Center for Radiation Research, an Office of Meas- urement Services and the following divisions: Applied Mathematics — Electricity — Mechanics — Heat — Optical Physics — Nuclear Sciences" — Applied Radiation 2 — Quantum Electronics 1 — Electromagnetics 3 — Time 3 1 1 and Frequency — Laboratory Astrophysics — Cryogenics . -

Carbon Dioxide Adsorption by Metal Organic Frameworks (Synthesis, Testing and Modeling)
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 8-8-2013 12:00 AM Carbon Dioxide Adsorption by Metal Organic Frameworks (Synthesis, Testing and Modeling) Rana Sabouni The University of Western Ontario Supervisor Prof. Sohrab Rohani The University of Western Ontario Graduate Program in Chemical and Biochemical Engineering A thesis submitted in partial fulfillment of the equirr ements for the degree in Doctor of Philosophy © Rana Sabouni 2013 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Other Chemical Engineering Commons Recommended Citation Sabouni, Rana, "Carbon Dioxide Adsorption by Metal Organic Frameworks (Synthesis, Testing and Modeling)" (2013). Electronic Thesis and Dissertation Repository. 1472. https://ir.lib.uwo.ca/etd/1472 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. i CARBON DIOXIDE ADSORPTION BY METAL ORGANIC FRAMEWORKS (SYNTHESIS, TESTING AND MODELING) (Thesis format: Integrated Article) by Rana Sabouni Graduate Program in Chemical and Biochemical Engineering A thesis submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy The School of Graduate and Postdoctoral Studies The University of Western Ontario London, Ontario, Canada Rana Sabouni 2013 ABSTRACT It is essential to capture carbon dioxide from flue gas because it is considered one of the main causes of global warming. Several materials and various methods have been reported for the CO2 capturing including adsorption onto zeolites, porous membranes, and absorption in amine solutions. -

Investigating the Inhibition Mechanism of L,D-Transpeptidase 5 from Mycobacterium Tuberculosis Using Computational Methods
INVESTIGATING THE INHIBITION MECHANISM OF L,D-TRANSPEPTIDASE 5 FROM MYCOBACTERIUM TUBERCULOSIS USING COMPUTATIONAL METHODS BY: GIDEON FEMI TOLUFASHE 216076453 Submitted in fulfilment of the requirements for the degree of Doctor of Philosophy in Pharmaceutical Chemistry School of Health Sciences, College of Health Sciences, University of KwaZulu-Natal, Durban, South Africa. 2018 PREFACE The work described in this thesis was conducted at the Catalysis and Peptide Research Unit, Westville Campus, University of KwaZulu-Natal, Durban, South Africa, under the supervision of Dr Bahareh Honarparvar, Prof. H.G. Kruger and Dr G.E.M. Maguire. This work has not been submitted in any form for any degree or diploma to any institution, where use has been made of the work of others, it is duly acknowledged in the text. Supervisors: Dr B. Honarparvar Date 30/10/2018 Prof. H. G. Kruger________________ Date ___________ Dr. G.E.M Maguire_______________ Date ___________ As candidate’s supervisor I agree to the submission of this thesis. i DECLARATION DECLARATION I- PLAGIARISM I, Gideon Femi Tolufashe declare that (i). The research reports in this thesis, except where otherwise indicated, is my original work. (ii). This thesis has not been submitted for any degree or examination at any other university. (iii). This thesis does not contain other person’s data, pictures, graphs or other information, unless specifically acknowledged as being sourced from other persons. (iv). This thesis does not contain other person’s writing, unless specifically acknowledged as being sourced from other researchers. Where other written sources have been quoted, then: a. Their words have been re-written, but the general information attributed to them has been referenced. -

Financial Statements and Trustees' Report 2017
Royal Society of Chemistry Financial Statements and Trustees’ Report 2017 About us Contents We are the professional body for chemists in the Welcome from our president 1 UK with a global community of more than 50,000 Our strategy: shaping the future of the chemical sciences 2 members in 125 countries, and an internationally Chemistry changes the world 2 renowned publisher of high quality chemical Chemistry is changing 2 science knowledge. We can enable that change 3 As a not-for-profit organisation, we invest our We have a plan to enable that change 3 surplus income to achieve our charitable objectives Champion the chemistry profession 3 in support of the chemical science community Disseminate chemical knowledge 3 and advancing chemistry. We are the largest non- Use our voice for chemistry 3 governmental investor in chemistry education in We will change how we work 3 the UK. Delivering our core roles: successes in 2017 4 We connect our community by holding scientific Champion for the chemistry profession 4 conferences, symposia, workshops and webinars. Set and maintain professional standards 5 We partner globally for the benefit of the chemical Support and bring together practising chemists 6 sciences. We support people teaching and practising Improve and enrich the teaching and learning of chemistry 6 chemistry in schools, colleges, universities and industry. And we are an influential voice for the Provider of high quality chemical science knowledge 8 chemical sciences. Maintain high publishing standards 8 Promote and enable the exchange of ideas 9 Our global community spans hundreds of thousands Facilitate collaboration across disciplines, sectors and borders 9 of scientists, librarians, teachers, students, pupils and Influential voice for the chemical sciences 10 people who love chemistry. -

Former Fellows Biographical Index Part
Former Fellows of The Royal Society of Edinburgh 1783 – 2002 Biographical Index Part Two ISBN 0 902198 84 X Published July 2006 © The Royal Society of Edinburgh 22-26 George Street, Edinburgh, EH2 2PQ BIOGRAPHICAL INDEX OF FORMER FELLOWS OF THE ROYAL SOCIETY OF EDINBURGH 1783 – 2002 PART II K-Z C D Waterston and A Macmillan Shearer This is a print-out of the biographical index of over 4000 former Fellows of the Royal Society of Edinburgh as held on the Society’s computer system in October 2005. It lists former Fellows from the foundation of the Society in 1783 to October 2002. Most are deceased Fellows up to and including the list given in the RSE Directory 2003 (Session 2002-3) but some former Fellows who left the Society by resignation or were removed from the roll are still living. HISTORY OF THE PROJECT Information on the Fellowship has been kept by the Society in many ways – unpublished sources include Council and Committee Minutes, Card Indices, and correspondence; published sources such as Transactions, Proceedings, Year Books, Billets, Candidates Lists, etc. All have been examined by the compilers, who have found the Minutes, particularly Committee Minutes, to be of variable quality, and it is to be regretted that the Society’s holdings of published billets and candidates lists are incomplete. The late Professor Neil Campbell prepared from these sources a loose-leaf list of some 1500 Ordinary Fellows elected during the Society’s first hundred years. He listed name and forenames, title where applicable and national honours, profession or discipline, position held, some information on membership of the other societies, dates of birth, election to the Society and death or resignation from the Society and reference to a printed biography. -

(8 PAGES) Subject Index, Vol:9, P.429, 1986
SUBJECT INDEX 1955-1985 moat-cited works 355 aatronomy 1961-1982 most-cited articles 55,118 1964 most-cited articles on 374 1976-1983 250,361 review articlea on 147 1963 most-citad articles, chemistry 413 Atlas of Science 1964 moat-citad articles planning for 41,44,46 Iife sciences 369 Attachment and Loss 35 physical acience 366 authora 1964 most-cited primary authors 346,355 name ordering on papers 353 1984 208,221 number of 393 1985 Nobel Prize awards and prizes, NAS Award for Excellence chemistry 336 in Scientific Reviewing 146 economics 307 literature 307,312 medicine 282 physics 336,340 1986 NAS Award for Excellence in Baruch, Bernard 114 Scientific Reviewing 146 La Bataille de Pharsale 314 The Battle of Pharsalus 314 A Beckett, Samuel 218 Beilstein, Friedrich 44 Benitez Sanchez, Jose 160 AIDS research, French and American Berzelius, Jons Jakob 1 contributions 347,391 bibliographies, editing with Sci-Mate Alice’s Adventuresin Wonderland 296 Editor 81 Allen, Gordon 43 The Biochemists’ Songbook 50 allergies BioGraffiti: A Natural Selection 49 effect of breaat feeding on 181 biomedical decision-making, role of effect of stress on 141 hospital library in 197,202 Analysis of Secretarial Duties and biomedical education curricula 27 Traits 114 Block, Konrad E. 282 animal studies, unreliability of transferring Bohr, Niels 6 results to humana 407 A Book of Science Verse 53 Annual Review of Physical Chemistry 9 breast feeding Arrhenius, Svante August 2 factors affecting choice of 165 art merits of mother’s milk 155 at ISI 130,176,228 Breastfeeding Handfwok 162 commissioned forlSI’s Caring Canter for Breastfeedirrg: A Guide for the Medical Children and Parents 260 Profession 156 role of turtle in 293 Brown, Michael S. -

1 CURRICULUM VITAE RUDOLPH A. MARCUS Personal Information
CURRICULUM VITAE RUDOLPH A. MARCUS Personal Information Date of Birth: July 21, 1923 Place of Birth: Montreal, Canada Married: Laura Hearne (dec. 2003), 1949 (three sons: Alan, Kenneth, and Raymond) Citizenship: U.S.A. (naturalized 1958) Education B.Sc. in Chemistry, McGill University, Montreal, Canada, 1943 Ph.D. in Chemistry, McGill University, 1946 Professional Experience Postdoctoral Research, National Research Council of Canada, Ottawa, Canada, 1946-49 Postdoctoral Research, University of North Carolina, 1949-51 Assistant Professor, Polytechnic Institute of Brooklyn, 1951-54; Associate Professor, 1954-58; Professor, 1958-64; (Acting Head, Division of Physical Chemistry, 1961-62) Member, Courant Institute of Mathematical Sciences, New York University, 1960-61 Professor, University of Illinois, 1964-78 (Head, Division of Physical Chemistry, 1967-68) Visiting Professor of Theoretical Chemistry, IBM, University of Oxford, England, 1975-76 Professorial Fellow, University College, University of Oxford, 1975-76 Arthur Amos Noyes Professor of Chemistry, California Institute of Technology, 1978-2012 Professor (hon.), Fudan University, Shanghai, China, 1994- Professor (hon.), Institute of Chemistry, Chinese Academy of Sciences, Beijing, China, 1995- Fellow (hon.), University College, University of Oxford, 1995- Linnett Visiting Professor of Chemistry, University of Cambridge, 1996 Honorable Visitor, National Science Council, Republic of China, 1999 Professor (hon.), China Ocean University, Qingdao, China, 2002 - Professor (hon.), Tianjin University, Tianjin, China, 2002- Professor (hon.) Dalian Institute of Chemical Physics, Dalian, China, 2005- Professor (hon.) Wenzhou Medical College, Wenzhou, China, 2005- Distinguished Affiliated Professor, Technical University of Munich, 2008- Visiting Nanyang Professor, Nanyang Institute of Technology, Singapore 2009- Chair Professor (hon.) University System of Taiwan, 2011 Distinguished Professor (hon.), Tumkur University, India, 2012 Arthur Amos Noyes Professor of Chemistry, California Institute of Technology, 1978-2013 John G. -

New Journal and Database Subscriptions – 2012 -2013
NEW JOURNAL AND DATABASE SUBSCRIPTIONS – 2012 -2013 New Journals Afterall: A Journal of Art, Context and Enquiry American Biology Teacher American Journal of Bioethics American Political Thought Annals of Tourism Research Art Documentation Biodiversity and Conservation Biomaterials Science BioScience Boom: A Journal of California California Archaeology California Management Review Catalysis Science & Technology Chemical Hazards in Industry China Journal Classical Antiquity Classical Philology Crime and Justice Critical Review of International Social and Political Philosophy Education in Chemistry Educational Technology Research Development Elephant Ethics Federal Sentencing Reporter Food & Function Frankie Gastronomica: The Journal of Food and Culture Haaretz Historical Studies in the Natural Sciences HOPOS: The Journal of the International Society for the History of Philosophy of Science Huntington Library Quarterly Indian Country Today Indonesia Journal Information, Communication & Society Innovation Policy and the Economy Integrative Biology Issues in Environmental Science and Technology Journal of Applied Remote Sensing Journal of Digital Media Management Journal of Empirical Research on Human Research Ethics Journal of Environmental Studies and Sciences Journal of Human Capital Journal of Labor Economics Journal of Leisure Research Journal of Micro/Nanolithography, MEMS, and MOEMS Journal of Modern History Journal of Nanophotonics Journal of North African Studies Journal of Palestine Studies Journal of Photonics for Energy Journal -

1- Report of Dr. Lee's Professor of Chemistry for the Year Ended 31
-1- REPORT OF DR. LEE’S PROFESSOR OF CHEMISTRY FOR THE YEAR ENDED 31st July 2003 Over the past year, several members of the Department were honoured in different ways, and we extend our warm congratulations to them all. We are delighted with the election of Professor John Brown to Fellowship of the Royal Society. Professor David Clary was elected to Honorary Membership of the American Academy of Arts and Sciences, as well as being elected a Fellow of the American Association for the Advancement of Science. In addition, he was elected to the Council of the Royal Society, and was awarded the Polanyi Medal of the Gas Kinetics Discussion Group of the Royal Society of Chemistry. Professor Richard Compton was nominated visiting Professor at the University of Săo Paulo in Brazil over the last summer. Professor Jacob Klein was awarded the Prize Lecture of the Colloid and Interface Division of the Japanese Chemical Society, while Professor Paul Madden was awarded the Statistical Mechanics and Simulation Industrial Award of the Royal Society of Chemistry sponsored by Unilever. Professor Richard Wayne was honoured by being chosen as the Hauptvortrag speaker at the 10th Fachbereichstag at the University of Wuppertal. Of our junior members, we note with pleasure that Jay Wadhawan, a 3rd year D.Phil. in Richard Compton’s group, was awarded a two-year Study Abroad Studentship by the Leverhulme Trust, while Susan Perkin, working in Jacob Klein’s group, was elected to a Jowett Senior Scholarship at Balliol. We were sorry to mark the death in September 2002 of Dr. -

GUT RSC Journals List
SCHEDULE A Publisher Content Section A Customer has access to the electronic versions of the following journals via an External route: Access Post - Copyright Journals E-ISSN years cancellation Owner* during Term access Analyst 1364-5528 2008-2018 2012-2018 RSC Analytical Methods 1 1759-9679 2009-2018 2012-2018 RSC Annual Reports on the Progress of Chemistry, A 1460-4760 2008-2013 2012-2013 RSC B 1460 4779 2008-2013 2012-2013 RSC C 1460-4787 2008-2013 2012-2013 RSC Biomaterials Science 1 2047-4849 2013-2018 2016-2018 RSC Catalysis Science & Technology 1 2044-4761 2011-2018 2013-2018 RSC Chemical Communications 1364-548X 2008-2018 2012-2018 RSC Chemical Science 1, 2 2041-6539 2010-2014 2012-2014 RSC Chemical Society Reviews 1460-4744 2008-2018 2012-2018 RSC Chemistry World 1749-5318 2012-2016 2012-2016 RSC CrystEngComm 1466-8033 2008-2018 2012-2018 RSC Dalton Transactions 1477-9234 2008-2018 2012-2018 RSC Education in Chemistry 1749-5326 2012-2016 2012-2016 RSC Energy & Environmental Science 1 1754-5706 2008-2018 2012-2018 RSC Environmental Science: Nano 1 2051-8161 2014-2018 2016-2018 RSC Environmental Science: Processes & Impacts including 2050-7895 2013-2018 2013-2018 RSC Journal of Environmental Monitoring (1464-0333) 2008-2012 2012 Environmental Science: Water Research & Technology 1 2053-1419 2015-2018 2017-2018 RSC Faraday Discussions 1364-5498 2008-2018 2012-2018 RSC Food & Function 1 2042-650X 2010-2018 2012-2018 RSC Green Chemistry 1463-9270 2008-2018 2012-2018 RSC Inorganic Chemistry Frontiers 1 2052-1553 2014-2018 2017-2018 -
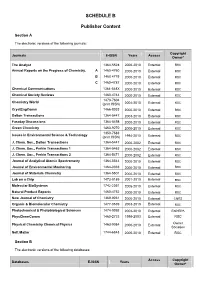
SCHEDULE B Publisher Content
SCHEDULE B Publisher Content Section A The electronic versions of the following journals: Copyright Journals E-ISSN Years Access Owner* The Analyst 1364-5528 2000-2010 External RSC Annual Reports on the Progress of Chemistry, A 1460-4760 2000-2010 External RSC B 1460 4779 2000-2010 External RSC C 1460-4787 2000-2010 External RSC Chemical Communications 1364-548X 2000-2010 External RSC Chemical Society Reviews 1460-4744 2000-2010 External RSC 1473-7604 Chemistry World (print ISSN) 2004-2010 External RSC CrystEngComm 1466-8033 2000-2010 External RSC Dalton Transactions 1364-5447 2003-2010 External RSC Faraday Discussions 1364-5498 2000-2010 External RSC Green Chemistry 1463-9270 2000-2010 External RSC 1350-7583 Issues in Environmental Science & Technology (print ISSN) 1994-2010 External RSC J. Chem. Soc., Dalton Transactions 1364-5447 2000-2002 External RSC J. Chem. Soc., Perkin Transactions 1 1364-5463 2000-2002 External RSC J. Chem. Soc., Perkin Transactions 2 1364-5471 2000-2002 External RSC Journal of Analytical Atomic Spectrometry 1364-5544 2000-2010 External RSC Journal of Environmental Monitoring 1464-0333 2000-2010 External RSC Journal of Materials Chemistry 1364-5501 2000-2010 External RSC Lab on a Chip 1473-0189 2001-2010 External RSC Molecular BioSystems 1742-2051 2005-2010 External RSC Natural Product Reports 1460-4752 2000-2010 External RSC New Journal of Chemistry 1369-9261 2000-2010 External CNRS Organic & Biomolecular Chemistry 1477-0539 2003-2010 External RSC Photochemical & Photobiological Sciences 1474-9092 2002-2010 -

1 Supporting Information Chemical Information Literacy at a Liberal Arts
Supporting Information Chemical Information Literacy at a Liberal Arts College George Greco Department of Chemistry Goucher College 1021 Dulaney Valley Road Baltimore, MD 21204 1 Table of Contents Course Syllabus 3-6 Handout 1: The Chemical Literature 7-8 Handout 2: Types of Papers 9 Handout 3: Citations 10-11 Handout 4: Secondary Sources 12-15 Handout 5: SciFinder 16-18 Handout 6: Free Public Databases 19-22 Handout 7: InChI and SMILES 23-25 Handout 8: Reaxys, Beilstein and Gmelin 26-27 Handout 9: Patents 28-32 Handout 10: Reading papers for current events 33 Handout 11: The publication process 34-37 Handout 12: Structure and sequence databases 38-41 Handout 13: Databases of Spectral and Thermodynamic Properties 42-43 HW #1: The Chemical Literature 44 SciFinder Assignment 45-46 HW #4: PubMed and PubChem 47 HW #5: SMILES and InChI 48 Current Events Assignment 49-50 Note about final exam 51 2 Chemistry 245 – Spring 2015 Chemical Information Literacy Instructor : Dr. George Greco Office : Hoffberger 220 Phone : 410-337-6313 Email : [email protected] Class Meetings : Tuesdays 11:45– 12:35 Hoffberger 223 Office hours: Monday 11:00-12:30, Wednesday 1:30-3:00 or by appointment (e-mail me) Optional Text : Chemical Information for Chemists: A Primer edited by Judith N. Currano and Dana L. Roth. Published by RSC Publishing. ISBN 978-1-84973-551-3 Disclaimer: This is the first time we are offering this course. It is my first time teaching a course like this. I don’t expect everything to go 100% smoothly.