Investigating the Inhibition Mechanism of L,D-Transpeptidase 5 from Mycobacterium Tuberculosis Using Computational Methods
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tufts Health Unify 202 0 List of Covered Drugs (Formulary)
Tufts Health Unify 202 0 List of Covered Drugs (Formulary) Effective: 12/01/2020 Tufts Health Plan P.O. Box 9194 Watertown, MA 02471-9194 Phone: 855.393.3154 Seven days a week, from 8 a.m. to 8 p.m. TuftsHealthUnify.org Formulary ID: 20533 Version: 20 H7419_6507B Approved DISCRIMINATION IS AGAINST THE LAW Tufts Health Plan complies with applicable Federal civil rights laws and does not discriminate on the basis of race, color, national origin, age, disability, or sex. Tufts Health Plan does not exclude people or treat them differently because of race, color, national origin, age, disability, or sex. Tufts Health Plan: . Provides free aids and services to people with disabilities to communicate effectively with us, such as: — Written information in other formats (large print, audio, accessible electronic formats, other formats) . Provides free language services to people whose primary language is not English, such as: — Qualified interpreters — Information written in other languages If you need these services, contact Tufts Health Plan Member Services at 855.393.3154. If you believe that Tufts Health Plan has failed to provide these services or discriminated in another way on the basis of race, color, national origin, age, disability, or sex, you can file a grievance with: Tufts Health Plan Attention: Civil Rights Coordinator, Legal Dept. 705 Mount Auburn St. Watertown, MA 02472 Phone: 888.880.8699 ext. 48000, [TTY number— 711 or 800.439.2370] Fax: 617.972.9048 Email: [email protected] You can file a grievance in person or by mail, fax, or email. If you need help filing a grievance, the Tufts Health Plan Civil Rights Coordinator is available to help you. -

PRIMAXIN (Imipenem and Cilastatin)
• Known hypersensitivity to any component of PRIMAXIN (4) HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use ----------------------- WARNINGS AND PRECAUTIONS ---------------------- PRIMAXIN safely and effectively. See full prescribing information • Hypersensitivity Reactions: Serious and occasionally fatal for PRIMAXIN. hypersensitivity (anaphylactic) reactions have been reported in patients receiving therapy with beta-lactams. If an allergic reaction PRIMAXIN® (imipenem and cilastatin) for Injection, for to PRIMAXIN occurs, discontinue the drug immediately (5.1). intravenous use • Seizure Potential: Seizures and other CNS adverse reactions, such Initial U.S. Approval: 1985 as confusional states and myoclonic activity, have been reported during treatment with PRIMAXIN. If focal tremors, myoclonus, or --------------------------- RECENT MAJOR CHANGES -------------------------- seizures occur, patients should be evaluated neurologically, placed Indications and Usage (1.9) 12/2016 on anticonvulsant therapy if not already instituted, and the dosage of Dosage and Administration (2) 12/2016 PRIMAXIN re-examined to determine whether it should be decreased or the antibacterial drug discontinued (5.2). ----------------------------INDICATIONS AND USAGE --------------------------- • Increased Seizure Potential Due to Interaction with Valproic Acid: PRIMAXIN for intravenous use is a combination of imipenem, a penem Co-administration of PRIMAXIN, to patients receiving valproic acid antibacterial, and cilastatin, a renal dehydropeptidase inhibitor, or divalproex sodium results in a reduction in valproic acid indicated for the treatment of the following serious infections caused by concentrations. The valproic acid concentrations may drop below designated susceptible bacteria: the therapeutic range as a result of this interaction, therefore • Lower respiratory tract infections. (1.1) increasing the risk of breakthrough seizures. The concomitant use of • Urinary tract infections. -

Western Sky Community Care Preferred Drug Locator Instructions: Preferred Drug List Includes a List of Drug Covered by Your Prescription 1
Preferred Drug List The Western Sky Community care Preferred Drug Locator Instructions: Preferred Drug List includes a list of 1. Within the PDF, click on the drug covered by your prescription Edit menu, then click Find. benefit. This list is updated often and may change. 2. In the Find box, type the name of To get the most up-to-date the medication you want to information, you may view the latest locate. Preferred Drug List on our website: WesternSkyCommunityCare.com 3. Click the Next button or call 1-844-543-8996 (TTY/TDD until you find the drug(s). 711). What is the Western Sky made to the drug list that may impact you. Community Care Preferred Drug List (PDL)? How do I use the Preferred Drug The preferred drug list (which is also List? called a “formulary”) is a list showing The best way to find your drug is by the drugs that can be covered by your going to the back of this book to the Western Sky Community Health Plan. index and looking it up by name. If the The drug listed will be covered as long drug is in all CAPITAL LETTERS (EX: as you: CIPRO TABS) the drug is a BRAND Have a medical need for the drug name drug and if the drug is in all lower case letters (ex: ciprofloxacin) Fill your drugs at a pharmacy the drug is a generic name drug. Next that we cover to your drug, you will see the page Follow any other rules that number where you can find coverage may apply to you as a member information. -

New Journal and Database Subscriptions – 2012 -2013
NEW JOURNAL AND DATABASE SUBSCRIPTIONS – 2012 -2013 New Journals Afterall: A Journal of Art, Context and Enquiry American Biology Teacher American Journal of Bioethics American Political Thought Annals of Tourism Research Art Documentation Biodiversity and Conservation Biomaterials Science BioScience Boom: A Journal of California California Archaeology California Management Review Catalysis Science & Technology Chemical Hazards in Industry China Journal Classical Antiquity Classical Philology Crime and Justice Critical Review of International Social and Political Philosophy Education in Chemistry Educational Technology Research Development Elephant Ethics Federal Sentencing Reporter Food & Function Frankie Gastronomica: The Journal of Food and Culture Haaretz Historical Studies in the Natural Sciences HOPOS: The Journal of the International Society for the History of Philosophy of Science Huntington Library Quarterly Indian Country Today Indonesia Journal Information, Communication & Society Innovation Policy and the Economy Integrative Biology Issues in Environmental Science and Technology Journal of Applied Remote Sensing Journal of Digital Media Management Journal of Empirical Research on Human Research Ethics Journal of Environmental Studies and Sciences Journal of Human Capital Journal of Labor Economics Journal of Leisure Research Journal of Micro/Nanolithography, MEMS, and MOEMS Journal of Modern History Journal of Nanophotonics Journal of North African Studies Journal of Palestine Studies Journal of Photonics for Energy Journal -

Appendix C Medication Tables
Appendix C Medication Tables Note: The medication tables are not meant to be inclusive lists of all available therapeutic agents. Approved medication tables will be updated regularly. Discrepancies must be reported. See Resource Section of this manual for additional contact information. Release Notes: Aspirin Table Version 1.0 Table 1.1 Aspirin and Aspirin-Containing Medications Acetylsalicylic Acid Acuprin 81 Alka-Seltzer Alka-Seltzer Morning Relief Anacin Arthritis Foundation Aspirin Arthritis Pain Ascriptin Arthritis Pain Formula ASA ASA Baby ASA Baby Chewable ASA Baby Coated ASA Bayer ASA Bayer Children's ASA Buffered ASA Children's ASA EC ASA Enteric Coated ASA/Maalox Ascriptin Aspergum Aspir-10 Aspir-Low Aspir-Lox Aspir-Mox Aspir-Trin Aspirbuf Aspircaf Aspirin Aspirin Baby Aspirin Bayer Aspirin Bayer Children's Aspirin Buffered Aspirin Child Aspirin Child Chewable Aspirin Children's Aspirin EC Aspirin Enteric Coated Specifications Manual for National Appendix C-1 Hospital Quality Measures Table 1.1 Aspirin and Aspirin-Containing Medications (continued) Aspirin Litecoat Aspirin Lo-Dose Aspirin Low Strength Aspirin Tri-Buffered Aspirin, Extended Release Aspirin/butalbital/caffeine Aspirin/caffeine Aspirin/pravachol Aspirin/pravastatin Aspirtab Bayer Aspirin Bayer Aspirin PM Extra Strength Bayer Children’s Bayer EC Bayer Enteric Coated Bayer Low Strength Bayer Plus Buffered ASA Buffered Aspirin Buffered Baby ASA Bufferin Bufferin Arthritis Strength Bufferin Extra Strength Buffex Cama Arthritis Reliever Child’s Aspirin Coated Aspirin -

GUT RSC Journals List
SCHEDULE A Publisher Content Section A Customer has access to the electronic versions of the following journals via an External route: Access Post - Copyright Journals E-ISSN years cancellation Owner* during Term access Analyst 1364-5528 2008-2018 2012-2018 RSC Analytical Methods 1 1759-9679 2009-2018 2012-2018 RSC Annual Reports on the Progress of Chemistry, A 1460-4760 2008-2013 2012-2013 RSC B 1460 4779 2008-2013 2012-2013 RSC C 1460-4787 2008-2013 2012-2013 RSC Biomaterials Science 1 2047-4849 2013-2018 2016-2018 RSC Catalysis Science & Technology 1 2044-4761 2011-2018 2013-2018 RSC Chemical Communications 1364-548X 2008-2018 2012-2018 RSC Chemical Science 1, 2 2041-6539 2010-2014 2012-2014 RSC Chemical Society Reviews 1460-4744 2008-2018 2012-2018 RSC Chemistry World 1749-5318 2012-2016 2012-2016 RSC CrystEngComm 1466-8033 2008-2018 2012-2018 RSC Dalton Transactions 1477-9234 2008-2018 2012-2018 RSC Education in Chemistry 1749-5326 2012-2016 2012-2016 RSC Energy & Environmental Science 1 1754-5706 2008-2018 2012-2018 RSC Environmental Science: Nano 1 2051-8161 2014-2018 2016-2018 RSC Environmental Science: Processes & Impacts including 2050-7895 2013-2018 2013-2018 RSC Journal of Environmental Monitoring (1464-0333) 2008-2012 2012 Environmental Science: Water Research & Technology 1 2053-1419 2015-2018 2017-2018 RSC Faraday Discussions 1364-5498 2008-2018 2012-2018 RSC Food & Function 1 2042-650X 2010-2018 2012-2018 RSC Green Chemistry 1463-9270 2008-2018 2012-2018 RSC Inorganic Chemistry Frontiers 1 2052-1553 2014-2018 2017-2018 -
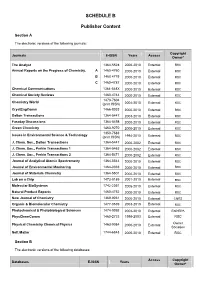
SCHEDULE B Publisher Content
SCHEDULE B Publisher Content Section A The electronic versions of the following journals: Copyright Journals E-ISSN Years Access Owner* The Analyst 1364-5528 2000-2010 External RSC Annual Reports on the Progress of Chemistry, A 1460-4760 2000-2010 External RSC B 1460 4779 2000-2010 External RSC C 1460-4787 2000-2010 External RSC Chemical Communications 1364-548X 2000-2010 External RSC Chemical Society Reviews 1460-4744 2000-2010 External RSC 1473-7604 Chemistry World (print ISSN) 2004-2010 External RSC CrystEngComm 1466-8033 2000-2010 External RSC Dalton Transactions 1364-5447 2003-2010 External RSC Faraday Discussions 1364-5498 2000-2010 External RSC Green Chemistry 1463-9270 2000-2010 External RSC 1350-7583 Issues in Environmental Science & Technology (print ISSN) 1994-2010 External RSC J. Chem. Soc., Dalton Transactions 1364-5447 2000-2002 External RSC J. Chem. Soc., Perkin Transactions 1 1364-5463 2000-2002 External RSC J. Chem. Soc., Perkin Transactions 2 1364-5471 2000-2002 External RSC Journal of Analytical Atomic Spectrometry 1364-5544 2000-2010 External RSC Journal of Environmental Monitoring 1464-0333 2000-2010 External RSC Journal of Materials Chemistry 1364-5501 2000-2010 External RSC Lab on a Chip 1473-0189 2001-2010 External RSC Molecular BioSystems 1742-2051 2005-2010 External RSC Natural Product Reports 1460-4752 2000-2010 External RSC New Journal of Chemistry 1369-9261 2000-2010 External CNRS Organic & Biomolecular Chemistry 1477-0539 2003-2010 External RSC Photochemical & Photobiological Sciences 1474-9092 2002-2010 -

1 Supporting Information Chemical Information Literacy at a Liberal Arts
Supporting Information Chemical Information Literacy at a Liberal Arts College George Greco Department of Chemistry Goucher College 1021 Dulaney Valley Road Baltimore, MD 21204 1 Table of Contents Course Syllabus 3-6 Handout 1: The Chemical Literature 7-8 Handout 2: Types of Papers 9 Handout 3: Citations 10-11 Handout 4: Secondary Sources 12-15 Handout 5: SciFinder 16-18 Handout 6: Free Public Databases 19-22 Handout 7: InChI and SMILES 23-25 Handout 8: Reaxys, Beilstein and Gmelin 26-27 Handout 9: Patents 28-32 Handout 10: Reading papers for current events 33 Handout 11: The publication process 34-37 Handout 12: Structure and sequence databases 38-41 Handout 13: Databases of Spectral and Thermodynamic Properties 42-43 HW #1: The Chemical Literature 44 SciFinder Assignment 45-46 HW #4: PubMed and PubChem 47 HW #5: SMILES and InChI 48 Current Events Assignment 49-50 Note about final exam 51 2 Chemistry 245 – Spring 2015 Chemical Information Literacy Instructor : Dr. George Greco Office : Hoffberger 220 Phone : 410-337-6313 Email : [email protected] Class Meetings : Tuesdays 11:45– 12:35 Hoffberger 223 Office hours: Monday 11:00-12:30, Wednesday 1:30-3:00 or by appointment (e-mail me) Optional Text : Chemical Information for Chemists: A Primer edited by Judith N. Currano and Dana L. Roth. Published by RSC Publishing. ISBN 978-1-84973-551-3 Disclaimer: This is the first time we are offering this course. It is my first time teaching a course like this. I don’t expect everything to go 100% smoothly. -

FDA Listing of Established Pharmacologic Class Text Phrases January 2021
FDA Listing of Established Pharmacologic Class Text Phrases January 2021 FDA EPC Text Phrase PLR regulations require that the following statement is included in the Highlights Indications and Usage heading if a drug is a member of an EPC [see 21 CFR 201.57(a)(6)]: “(Drug) is a (FDA EPC Text Phrase) indicated for Active Moiety Name [indication(s)].” For each listed active moiety, the associated FDA EPC text phrase is included in this document. For more information about how FDA determines the EPC Text Phrase, see the 2009 "Determining EPC for Use in the Highlights" guidance and 2013 "Determining EPC for Use in the Highlights" MAPP 7400.13. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. -

FEP® Blue Focus Formulary (907)
FEP® Blue Focus Formulary (907) Effective January 1, 2022 The FEP formulary includes a preferred drug list which is comprised of Tier 1, generics and Tier 2, preferred brand-name drugs, preferred generic specialty drugs, and preferred brand-name specialty drugs. Ask your physician if there is a generic drug available to treat your condition. If there is no generic drug available, ask your physician to prescribe a preferred brand-name drug. The preferred brand-name drugs within our formulary are listed to identify medicines that are clinically appropriate and cost-effective. Click on the category name in the Table of Contents below to go directly to that page INTRODUCTION ........................................................................................................................................................................................................................ 5 PREFACE ................................................................................................................................................................................................................................... 5 PRIOR APPROVAL ................................................................................................................................................................................................................... 5 QUANTITY LIMITATIONS ........................................................................................................................................................................................................ -

Perioperative Medication Management - Adult/Pediatric - Inpatient/Ambulatory Clinical Practice Guideline
Effective 6/11/2020. Contact [email protected] for previous versions. Perioperative Medication Management - Adult/Pediatric - Inpatient/Ambulatory Clinical Practice Guideline Note: Active Table of Contents – Click to follow link INTRODUCTION........................................................................................................................... 3 SCOPE....................................................................................................................................... 3 DEFINITIONS .............................................................................................................................. 3 RECOMMENDATIONS ................................................................................................................... 4 METHODOLOGY .........................................................................................................................28 COLLATERAL TOOLS & RESOURCES..................................................................................................31 APPENDIX A: PERIOPERATIVE MEDICATION MANAGEMENT .................................................................32 APPENDIX B: TREATMENT ALGORITHM FOR THE TIMING OF ELECTIVE NONCARDIAC SURGERY IN PATIENTS WITH CORONARY STENTS .....................................................................................................................58 APPENDIX C: METHYLENE BLUE AND SEROTONIN SYNDROME ...............................................................59 APPENDIX D: AMINOLEVULINIC ACID AND PHOTOTOXICITY